Alfaro Lara ER, Sánchez Pozo MI, Desongles Corrales T, Santos Rubio MD
Unidad de Gestión Clínica de Farmacia. Hospital Universitario Virgen del Rocío. Sevilla (España)
Resumen
Objetivos: Identificar los posibles riesgos, utilizando la metodología AMFE, en el área de ensayos clínicos de un hospital con el fin de mejorar la seguridad de los procedimientos habituales.
Métodos: Se formó un equipo multidisciplinar formado por dos enfermeras, dos coordinadores de ensayos, un monitor, un técnico de Farmacia y cuatro farmacéuticos. Se definieron claramente el proceso y los sub-procesos de estudio. Se realizó un diagrama de flujo del proceso y se identificaron y analizaron sus posibles modos de fallo.
Resultados: El proceso se dividió en ocho subprocesos: visita inicial, la recepción de las muestras, la prescripción, la custodia, preparación, dispensación, destrucción/devolución de muestras y visita de cierre. 24 de los 36 modos de fallo identificados fueron clasificados como de alto riesgo (NPR >100). El mayor número de modos de fallo potenciales se concentró en dos subprocesos: preparación de muestras y dispensación. Los modos de fallo que tuvieron una mayor reducción del riesgo después de la aplicación de la medida de mejora fueron: información incorrecta verbal durante la visita inicial, ubicación incorrecta, preparación de muestras errónea, esquema mal diseñado en el programa de citostáticos y confusión de pacientes. Todas las recomendaciones establecidas, excepto una, han sido ya implementadas.
Conclusiones: La metodología AMFE es una herramienta útil cuando se aplica al área de ensayos clínicos. Su aplicación ha permitido reducir el riesgo de forma significativa. La implementación de las medidas recomendadas ha reducido el NPR, siendo inferior a 100 tras el establecimiento de las recomendaciones.
Palabras clave: Análisis modal de fallos y efectos, seguridad, ensayos clínicos, calidad.
Analysis of failures and effects applied to the area of clinical trials
SUMMARY
Objectives: To identify the risks in the clinical trials area using the FMEA methodology in order to improve the safety of the usual procedures.
Methods: A multidisciplinary team consisting of two nurses, two study coordinators, one clinical research associate, one pharmacy technician and four pharmacists was assembled. The process and sub-processes were clearly defined. A flow diagram of the process was made and potential failure modes were identified and evaluated.
Results: The process was divided in eight sub-processes: initial visit, receipt of samples, prescription, custody, preparation, dispensation, sample destruction/devolution and site closeout visit. Twenty four out of the 36 identified failure modes were classified as high risk (RPN >100). The largest number of potential failure modes is concentrated in two sub-processes: sample preparation and dispensation. The failure modes that had a greater risk reduction after the improvement measure implementation were: incorrect verbal information during initial visit, incorrect location, erroneous sample preparation, wrong design schema in the cytostatics program, mixing up patients. All established recommendations except one have been already implemented.
Conclusions: The FMEA methodology was a useful tool when applied to the area of clinical trials. It allowed us to reduce the risk significantly. The implementation of the recommended measures has reduced the RPN, being less than 100 after the establishment of the recommendations.
Key Words: Modal analysis of failures and effects, safety, clinical trials, quality.
____
INTRODUCCIÓN
La seguridad en los procesos asistenciales es una preocupación creciente para los sistemas de salud. Las razones para el alto índice de errores potenciales son, en general, la incertidumbre de los efectos de los medicamentos, la complejidad de los procesos, la falta de estandarización, la falta de información y la propia naturaleza humana.
Los ensayos clínicos (EC) con medicamentos, desde el punto de vista de la seguridad, presentan estrictos protocolos de trabajo y criterios de actuación muy definidos, requiriendo una atención, formación y seguimiento máximo por parte de los responsables. Además, la información es muy limitada, desconociendo qué medicamento se administra, las propiedades del mismo y siendo muy complicado acceder a esta información por los profesionales de la salud que no pertenecen al equipo investigador1.
Dados los continuos avances en investigación clínica, cada vez es mayor el número de ensayos clínicos activos en los centros hospitalarios, así como el grado de complejidad de éstos. También este incremento se ha visto acompañado de una mayor participación activa del farmacéutico responsable en el desarrollo de los EC en el hospital, encontrándose entre sus actividades habituales, la información al paciente durante la dispensación de las muestras de investigación clínica (MIC), al personal de enfermería, y demás miembros del equipo investigador cuando proceda, aleatorización del paciente incluido, asignación de las muestras, preparación… y todo el apoyo logístico, tanto a investigadores como al promotor, que garantiza la utilización correcta de los medicamentos del EC2. Sin embargo, la descripción de estos procesos en la Farmacia no se suele incluir en los protocolos de los EC, dependiendo de los Procedimientos Normalizados de Trabajo del área de EC dentro los Servicio de Farmacia Hospitalarios (SFH).
El área de ensayos clínicos dentro de los SFH es un área especialmente vulnerable a los errores de medicación por distintos motivos: el circuito que sigue la medicación de EC es complejo y totalmente diferente al resto de medicación hospitalaria, son múltiples profesionales los que intervienen durante el curso de un EC, lo cual requiere una actividad coordinada, cualquier mínimo desvío del protocolo puede tener graves repercusiones, coexisten diferentes MIC de un mismo fármaco experimental por pertenecer a diferentes ensayos, los códigos de protocolo no facilitan la identificación del mismo, pudiendo requerir otro sistema de identificación propio del SFH paralelo, cada protocolo de ensayo requiere unas condiciones de actuación específicas, etc.
Las Normas de Buena Práctica Clínica3 señalan la necesidad de establecer sistemas con procedimientos que aseguren la calidad de todos los aspectos del ensayo clínico. Debemos pues establecer medidas y actuaciones que mejoren la seguridad del paciente, para reducir la probabilidad de eventos adversos relacionados con el EC y las desviaciones de protocolo.
El análisis modal de fallos y efectos (AMFE) es un instrumento analítico recomendado por el Institute of Medicine (IOM) y la Joint Commission on Accreditation of Healthcare Organizations (JCAHO) (Standard LD.5.2), como procedimiento idóneo para lograr la seguridad en los procesos sanitarios. Muchos sectores industriales como los aeroespaciales y de la automoción lo están empleando desde hace décadas, aunque en la sanidad no se comenzó su aplicación hasta la década de los noventa con su adaptación a este ámbito por el Departamento de Veteranos de EEUU4. El AMFE es un instrumento preventivo que reduce los riesgos para los usuarios de un producto o servicio, tanto en las fases del diseño como en las fases de producción. Por ejemplo, en la industria aeroespacial, el AMFE puede prevenir que un avión tenga defectos en su diseño o fabricación para que no ocurran accidentes por defectos en el aparato.
Esta metodología se ha utilizado en campos complejos y de alto riesgo como la oncología pediátrica, los cuidados críticos o la radioterapia5-7. La aplicación de esta metodología al ámbito sanitario en España es muy reciente y existen pocas publicaciones al respecto8-11.
El principal objetivo de nuestro estudio es la identificación de riesgos en el área de ensayos clínicos, en base a todas las actividades que implica al SFH, de forma proactiva, su priorización y el establecimiento de medidas de mejora en la seguridad de los procedimientos utilizados.
MÉTODOS
El Hospital Universitario Virgen del Rocío es un hospital público que cuenta con la cartera de servicios más amplia para una población básica asignada de medio millón de habitantes en la provincia de Sevilla, siendo en algunas de sus especialidades más complejas, hospital de referencia para toda la Comunidad Autónoma Andaluza. Actualmente el centro tiene 358 ensayos clínicos activos, tratándose, la gran mayoría de MIC, de fármacos citostáticos.
La dispensación de las MIC se realiza en todos los casos a través del SFH. En caso de MIC para administración por vía oral o intravenosa (excluyendo citostáticos), se dispensa a través de una formulario específico que actúa como “Prescripción de medicamentos de ensayos clínicos” al paciente, y en algunos casos, al equipo investigador. En las MIC citostáticas administradas por vía intravenosa, la prescripción es transcrita y validada por el farmacéutico al programa informático que gestiona dicha medicación. Tras este paso, el técnico de Farmacia de EC prepara los viales a dispensar según la “hoja de preparación” obtenida y el farmacéutico de EC revisa dicha dispensación. Finalmente, enfermería prepara las dosis prescritas según instrucciones facilitadas por el farmacéutico y se administra al paciente en Hospital de Día12.
El AMFE ha sido llevado a cabo en un total de 2 meses. Las acciones de mejora fueron implementadas durante un período de 4 meses.
Composición del equipo multidisciplinar
Siguiendo la metodología AMFE, el primer paso fue constituir un equipo multidisciplinar. En el grupo de trabajo se incluyeron las siguientes figuras:
- Asesor de Calidad y experto en metodología AMFE.
- Coordinador de grupo encargado de la organización de las reuniones y de garantizar el correcto funcionamiento del equipo.
- Personas con experiencia y conocimientos de la unidad y del procedimiento objeto del AMFE, incluyendo para ello a cada uno de los diferentes profesionales que intervienen en el curso del EC:
- 2 enfermeras que habitualmente preparan MIC citostáticas, una con más experiencia y otra menos experimentada, para que cada una pudiese dar un enfoque diferente.
- 2 coordinadoras de estudio del servicio de Oncología, igualmente una más experimentada y otra de menor experiencia.
- 1 Clinical Research Associate (CRA).
- 1 técnico de Farmacia del área de ensayos clínicos.
- 4 farmacéuticas, 3 de ellas hospitalarias, siendo: una la experta en la metodología, otra la responsable final de dispensación de medicación a pacientes externos y de ensayos clínicos, y dos, las farmacéuticas responsables de EC en la práctica habitual.
Uno o varios de los anteriores roles podían recaer en un mismo miembro del equipo, de forma que el grupo estuvo formado por un total de 10 personas.
Proceso AMFE: área de ensayos clínicos del SFH
Se estableció una definición del proceso a evaluar y una descripción de los componentes. Este paso permite una mayor comprensión del proceso a evaluar por parte del equipo. Consiste en delimitar claramente el proceso y describir todos y cada uno de los subprocesos que lo integran. En la definición de este proceso fueron claves las aportaciones del personal propio de la unidad, puesto que se trata de aquellas actividades que implican de cualquier modo al SFH. Para facilitar esta tarea se simplificó el proceso y se dividió en sus respectivos subprocesos fundamentales.
Al tratarse de un proceso complejo, para facilitar el trabajo, se analizaron de forma independiente cada uno de los subprocesos descritos en el diagrama de flujo. Para cada uno de ellos, se identificaron los diferentes modos de fallo potenciales, entendiéndose por modo de fallo potencial toda forma en que es posible que un servicio o proceso falle. Del mismo modo, se identificaron las causas que podían originar los diferentes modos de fallo, así como los efectos que podrían tener en el paciente y/o sistema en caso de producirse. Para esta tarea se utilizó la técnica del “brainstorming”, en su versión oral, que se desarrolló en diferentes fases:
- Fase de generación de ideas: en la que cada miembro del grupo aportó sus ideas sin ningún tipo de censura por parte del resto del equipo.
- Fase de clarificación: con la que se pretendía garantizar que todos los miembros del equipo comprendían claramente cada una de las aportaciones.
- Fase de evaluación: llevada a cabo para eliminar las repeticiones y aportaciones fuera del ámbito tratado, así como para agrupar aportaciones afines.
Para calcular el impacto asociado a cada modo de fallo potencial se utilizó el Número de Prioridad de Riesgo (NPR), obtenido mediante una ecuación en la que intervienen tres variables:
– GRAVEDAD o impacto en el paciente y/o en el servicio (G).
– OCURRENCIA o probabilidad de ocurrir (O).
– DETECTABILIDAD o probabilidad de detectarlo en caso de ocurrir (D).
A continuación se muestra la ecuación que permite obtener este Número de Prioridad de Riesgo:

Para asignar una puntuación numérica a la severidad, probabilidad de ocurrir y capacidad de detección de cada uno de los modos de fallo identificados se usaron las escalas de valoración de gravedad, ocurrencia y detectabilidad proporcionadas por el Ministerio de Sanidad y Consumo (Tabla 1) que contemplan una puntuación de 1 a 10 para cada una de las variables.

Se establecieron medidas de mejora para todos los modos de fallo identificados cuyo resultado de NPR sobrepasó el valor 100, por tratarse de puntos de especial riesgo. Se calculó para estos puntos críticos el porcentaje de reducción de NPR y se consideró que las medidas de mejora serían útiles en aquellos con una reducción teórica por encima del 50%. Estas medidas de mejora se incorporaron al proceso finalizando así el rediseño del mismo. Se calculó el NPR acumulado como el sumatorio de los NPR.
RESULTADOS
Se realizaron un total de 6 reuniones presenciales por parte de los miembros del equipo formado. En la primera de ellas, la experta en la metodología expuso una sesión introductoria de 1 h sobre el desarrollo del análisis. La duración media de cada reunión fue aproximadamente de 2,5 h.
El proceso fue dividido en 8 subprocesos: visita de inicio, recepción de muestras, prescripción, custodia, preparación, dispensación, destrucción/devolución de MIC y visita de cierre (Figura 1).

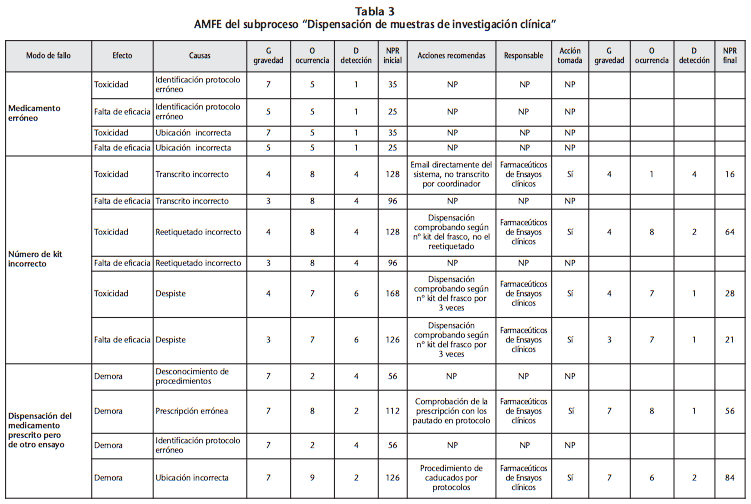
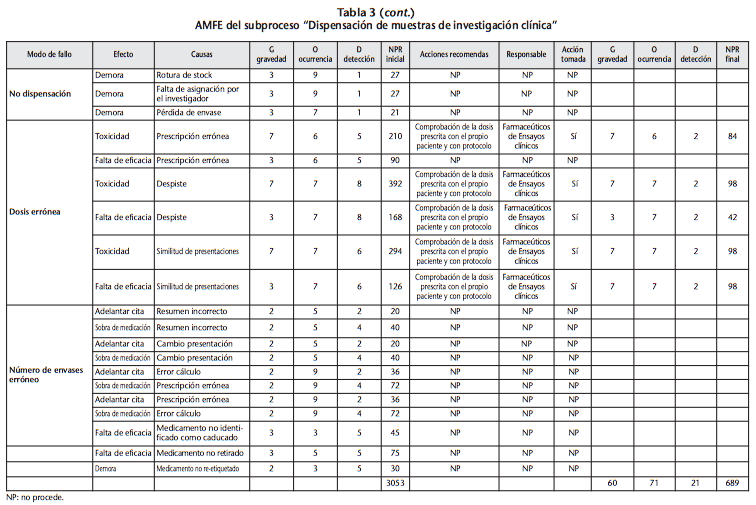
Se identificaron un total de 36 modos de fallo, con 61 causas asociadas y con efectos cuya gravedad varió entre 1 y 9. Para cada uno de ellos se consensuó el efecto, las posibles causas, el NPR inicial, la acción recomendada, y el NPR final (teórico). El NPR inicial acumulado fue de 6669 (rango: 4-392) y tras las acciones recomendadas el NPR final acumulado fue de 3711 (rango: 4-112). Se obtuvieron puntuaciones de NPR por encima de 100 en 24 de los modos de fallo, considerados críticos. El subproceso con más modos de fallos con NPR >100 fue el de dispensación de las muestras de EC, con un total de 10, y sólo los subprocesos de destrucción/devolución de muestras y visita de cierre estuvieron exentos de puntos críticos (NPR >100).
El mayor número de potenciales modos de fallo se concentra en dos subprocesos: preparación de las muestras y su dispensación, con unos NPR iniciales totales de 1264, en el primer caso, y 3053, en el segundo. El modo de fallo considerado de mayor riesgo (NPR=360) durante la preparación fue el de “introducir el esquema quimioterápico del ensayo en el programa de preparación de citostáticos de forma incorrecta”, por un mal entendimiento del protocolo o un mal dominio del programa, lo que podía producir toxicidad en el paciente. La acción de mejora establecida fue la de que el promotor tuviese que validar dicho esquema una vez introducido su diseño en el programa de preparación, tras la visita de inicio, y previo a la inclusión del primer paciente. Ello permitía reducir el NPR a 56. El modo de fallo considerado de mayor riesgo (NPR=392) en el momento de la dispensación fue el de dispensar una muestra del medicamento prescrito pero a una dosis superior por despiste, que podía ocasionar toxicidad en el paciente. La acción de mejora establecida fue verificar siempre previo a la dispensación la dosis que se ha prescrito con el propio paciente y con lo que establece el protocolo. Ello permitía reducir el NPR a 98.
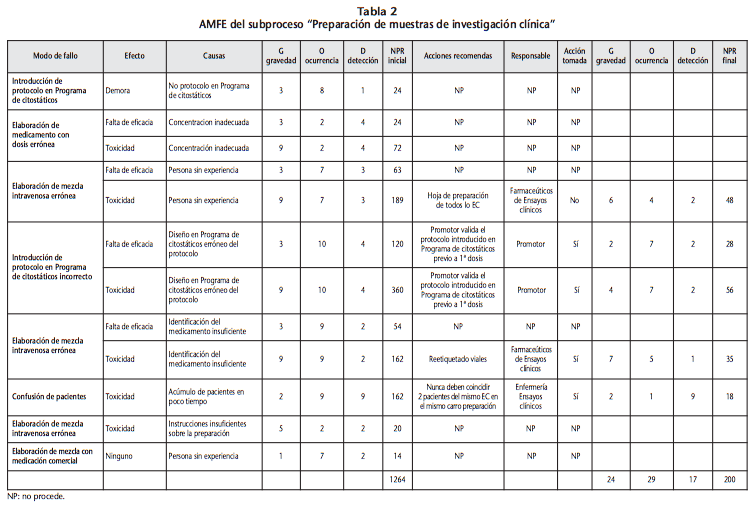
Los modos de fallo, así como sus causas, efectos, NPR estimados al inicio y tras la implantación teórica de la acción de mejora, así como las recomendaciones de mejora pueden apreciarse para ambos subprocesos en las tablas 2 y 3.
Los modos de fallo que sufrieron una mayor reducción del riesgo tras la implantación de la medida de mejora, por subproceso, fueron (ver Tablas 2 y 4):
- Visita de inicio: para “información verbal incorrecta” a causa de un monitor desinformado, el NPR descendió de 160 a 36 si Farmacia solicita la información de forma previa y por escrito.
- Custodia: en casos de “ubicación incorrecta” por despiste, el NPR disminuye de 288 a 84 tras establecer revisiones externas periódicas por la responsable del área.
- Preparación: para la “elaboración errónea de la muestra” por personal inexperto, el NPR disminuyó de 189 a 48 tras elaborar hojas de preparación específicas para la medicación de ensayo dirigidas a enfermería.
- Preparación: para un “diseño de esquema incorrecto en el programa de citostáticos”, el NPR disminuyó de 360 a 56 si el promotor valida dicho esquema previo al inicio del ensayo.
- Preparación: en caso de “confusión de pacientes” porque se acumulen distintos pacientes del mismo ensayo en poco tiempo, el NPR disminuyó de 162 a 18 tras instaurar que el técnico nunca prepare en el mismo carro muestras de distintos pacientes de un mismo ensayo.
Todas las recomendaciones establecidas, salvo una, han sido implementadas en el área de ensayos clínicos. Entre ellas destacan: que la información recibida durante la visita de inicio se obtenga de forma escrita a través de un formulario diseñado a tal fin, la validación por el promotor del esquema de tratamiento del ensayo tras su transcripción al programa de citostáticos, la realización de auditorias internas periódicas para verificar el correcto seguimiento del procedimientos estandarizado sobre la custodia de las muestras o el reetiquetado de las muestras cuya etiqueta pudiese generar confusión. La única recomendación aun no implantada es la de diseñar, para cada uno de los ensayos clínicos cuyas muestra/s requieren preparación por enfermería, una hoja de instrucciones específicas sobre la preparación de la muestra en cuestión.
DISCUSIÓN
La metodología AMFE aplicada a todos los subprocesos que tienen lugar en el área de ensayos ha permitido disminuir el riesgo de forma importante, mostrándose como una herramienta útil para el análisis proactivo de dichos riesgos. La implementación de las acciones de mejora recomendadas ha conseguido reducir el NPR acumulado de forma importante, lo cual también se observa en que la gran mayoría de modos de fallo considerados críticos, pasan a tener NPR <100 tras el establecimiento de las recomendaciones.
Los subprocesos de preparación y dispensación de las muestra de IC son los que entrañan un mayor número de puntos débiles. Algunos de los potenciales efectos contemplados en el subproceso de preparación fueron considerados de elevada gravedad (valores de G=9). En este sentido, hay que tener en cuenta que gran parte de las MIC que requieren preparación son fármacos citostáticos de administración intravenosa y cuyas dosis y efectos aún no están bien establecidos y, tal como recomienda la metodología, hay que situarse en el peor escenario posible, de aquí, valores de NPR tan elevados.
De la dispensación, destacan algunos modos de fallo con valores de ocurrencia también altos (8-9), lo que dispara también el NPR. El hecho de que estos posibles errores se puedan dar con más frecuencia que otros puede atribuirse al elevado número de ensayos clínicos activos en el centro (actualmente 358 ensayos), al elevado ritmo de reclutamiento en cada uno de los ensayos por tratarse de un hospital de tercer nivel con un papel muy activo en la investigación clínica, y a que la gran mayoría de dispensaciones se realiza de forma individualizada a cada paciente. Todo ello, unido además a la creciente complejidad en los protocolos de ensayo clínicos y a las peculiaridades de cada dispensación (asignación mediante sistema automatizado, acondicionamiento de las muestras, confirmación de los kits dispensados mediante sistema automatizado…).
Como se ha mencionado anteriormente, la utilización del método AMFE en el ámbito de la salud, es cada vez más frecuente. Sin embargo, y que los autores conozcan, este es el primer trabajo que se ha realizado en un área hospitalaria, dentro del servicio de Farmacia, de tanta envergadura y complejidad como es la investigación clínica con medicamentos. El constante aumento en número y complejidad de los estudios, así como la implicación por parte de personal interno (diferentes especialidades médicas, coordinadores de estudio, farmacéuticos…) y personal externo (promotor, monitor, pacientes…) durante el desarrollo del ensayo clínico, hace necesario un control exhaustivo de los procedimientos internos y una actitud proactiva para evitar o disminuir posibles errores, sobre todo en los que a preparación y dispensación de las muestras de investigación clínica se refiere.
Puesto que un AMFE depende de los miembros del grupo que examinan los fallos, sus resultados están limitados por la experiencia previa de los miembros. Sin embargo, esto es minimizado por la composición multidisciplinar del equipo13, así como por el hecho de que este método no se utiliza como herramienta única para valorar la seguridad y calidad de los procedimientos, sino que se complementa con un registro interno de incidentes y errores de medicación (según la terminología y clasificación de errores estandarizada por el grupo de la Sociedad Española de Farmacia Hospitalaria)14, como herramientas retrospectivas, así como con autoauditorías internas.
La metodología AMFE es una herramienta útil en la detección de riesgos potenciales en el área de ensayos clínicos, dentro del Servicio de Farmacia de un hospital de tercer grado, así como en el diseño e implementación de acciones de mejora que repercuten en la calidad del proceso global. Actualmente, esta metodología se está poniendo en marcha en otros departamentos del Servicio de Farmacia.

Conflicto de intereses: Los autores declaran no tener conflictos de intereses.
BIBLIOGRAFÍA
- Martínez Niet C. Ensayos clínicos en España. Madrid: Master Line and Prodigio SL, 2010.
- Quevedo de Torres A, Pérez Bravo L, Fernández Fernández A. Participación del farmacéutico de hospital en la investigación clínica. Farm Hosp. 1999;23:24-41.
- Guideline on Good Clinical Practice. EMA/CPMP/ ICH/135/95. [Internet] Agencia Europea del Medicamento (EMA); 2002. URL: http://www.ema.europa.eu/ docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdf.
- De Rosier J, Stalhandske E, Bagian JP, Nudell T. Using Health care failure mode and effect análisis: the VA National Center for Patiene Safety’s prospective risk anlysis system. Jt Comm J Qual Improv. 2002;28:248-67.
- Tilburg C, Leistikow I, Rademaker C, Bierings MB, Dijk A. Health care failure mode and effect analysis: a useful proactive risk analysis in a pediatric oncology ward. Qual Saf Health Care. 2006;15:58-64.
- Apkon M, Leonard J, Probst L, DeLizio L, Vitale R, et al. Design of a safer approach to intravenous drug infusions: failure mode effects analysis. Qual Saf Health Care. 2004;13:265-71.
- Govindarajan R, Molerob J, Tusetb V, Arellanob A, Ballesterb R,Cardenalc J, et al. El análisis modal de fallos y efectos (AMFE) ayuda a aumentar la seguridad en radioterapia. Rev Calid Asist. 2007;22:299-309.
- Palacio Lapuente F, Hernandez Galindo M, Amezqueta Goni C, Lapuente Heppe I, Sola Saravia C. Managing the atrial fibrillation process: an integral approach. Rev Calid Asist. 2013;28:19-27.
- Pérez Lázaro JJ, Fernández Ruiz I, Tejedor Fernández M, Guerra de Hoyos JA, Jiménez Rodríguez M, de Pazzis Die de Ortega M, et al. Identifying and prioritising adverse episodes and failures related to patient safety in Pain Treatment Units. Rev Esp Anestesiol Reanim. 2012;59:423-9.
- Cañada Dorado A, Cardenas Valladolid J, Espejo Matorrales F, Garcia Ferradal I, Sastre Paez S, Vicente Martin I. Improving the continuous care process in primary care during weekends and holidays: redesigning and FMEA. Rev Calid Asist. 2010;25:365-71.
- Alonso-Ovies A, Álvarez-Rodríguez J, del Mar García-Gálvez M, Velayos-Amo C, Balugo-Huertas S, Álvarez-Morales A. Usefulness of failure mode and effects analysis to improve patient safety during the process of incorporating new nurses in an intensive care unit. Med Clin (Barc). 2010;135 (Suppl 1):45-53.
- Procedimiento normalizado de trabajo: Documento para el monitor y/o promotor, versión 3. Área de ensayos clínicos de Farmacia. Hospital Universitario Virgen del Rocío (Sevilla).
- Sánchez-Muñoz LA, Mayor-Toranzo E, Rodríguez-Martín C. Análisis modal de fallos y efectos del sistema de utilización de medicamentos. Farm Hosp. 2012;36:299-300.
- Otero López MJ, Codina Jané C, Tamés Alonso MJ, Pérez Encinas M, en representación del grupo de trabajo Ruiz-Jarabo 2000. Errores de medicación: estandarización de la terminología y clasificación. Farm Hosp. 2003;27:137-49.
____
Descargar artículo en PDF: Análisis modal de fallos y efectos aplicado al área de ensayos clínicos
Artículo dentro del número: VOL. 25 – Nº3 – 2015
____