Sánchez-Azofra A1, Calvo-García A2, Ruiz-García S2, Girón Moreno RM1, Ibáñez Zurriaga MD2, Aldave Orzaiz B1, Pastor Sanz MT3, Ancochea Bermúdez J4, Morell Baladrón A5
1 Pulmonologist. Pneumology Department
2 Pharmacist. Pharmacy Department
3 Statistician. Pneumology Department
4 Head of Pneumology Department
5 Head of Pharmacy Department
La Princesa University Hospital. Madrid (Spain)
Fecha de recepción: 04/02/2020 – Fecha de aceptación: 18/02/2020
Correspondencia: Silvia Ruiz-García – Hospital Universitario de La Princesa (Departamento de Farmacia) – C/Diego de León, 62 – 28006 Madrid (España)
sruizg@salud.Madrid.org
_____
SUMMARY
Background: Cystic fibrosis (CF) is the most serious and frequent hereditary autosomal disease that causes respiratory, hepatic and pancreatic dysfunction. The aim of the study was to assess the pharmaceutical and medical cost in CF outpatients from the Adult Cystic Fibrosis Unit at third level hospital.
Material and methods: Retrospective observational study in adult CF patients throughout the year 2017. Demographic and clinical variables were included. All of the medical variables considered were directly related to the disease. Considered cost were laboratory selling price notified in Nomenclator. Medical costs were calculated based on laboratory’s price list and hospital medical procedures.
Results: 89 CF patients enter the study, and 57 patients were finally included. The mean age was 32.5 years, 56.1% were female. 36.5% patients were homozygous for Phe508del, 40.4% heterozygous, and 22.8% had another mutation. The average FEV1 was 72.2%. 33.3% patients were colonized by sensitive Pseudomonas aeruginosa (PA) and 7.0% by multidrug-resistant PA. Total costs per year was EUR 623,981.3, (87.6% drug costs and 12.4% medical costs).
Medical, drug and total costs were higher in Phe508del/Phe508del mutation group than Phe508del/other and other/other (p<0.05). Microbial colonization increased costs (p<0.05); colonized by sensitive PA had statistically significant higher drug and total costs, similar in multidrug resistant PA. Medical costs increase with severity level of lung function (p=0.001), also drug and total costs with the exception of severe patients.
CF is a relative costly disease for the healthcare system. In our study homozygous Phe508del mutation patients, lows values of FEV1 and colonization had higher cost.
Key words: Antibiotic, CFTR mutation, costs analysis, cystic fibrosis, FEV, Pseudomonas aeruginosa.
Tratamiento ambulatorio de fibrosis quística y costes médicos: un análisis retrospectivo
RESUMEN
Introducción: La fibrosis quística (FQ) es la enfermedad autosómica hereditaria más grave y frecuente que cursa con disfunción respiratoria, hepática y pancreática. El objetivo del estudio fue evaluar el coste farmacéutico y médico directo en pacientes ambulatorios de FQ de la Unidad de Fibrosis Quística de Adultos en un hospital de tercer nivel.
Material y métodos: Estudio observacional, retrospectivo, en pacientes adultos con FQ a lo largo del año 2017. Se recogieron variables demográficas y clínicas. Todas las variables médicas consideradas estaban directamente relacionadas con la enfermedad. Los costes considerados fueron los precios de venta de laboratorio notificado en Nomenclator. Los costes médicos se calcularon en base a la lista de precios del laboratorio y los procedimientos médicos hospitalarios.
Resultados: Se realizó el screening en 89 pacientes con FQ, y finalmente se incluyeron 57 pacientes. La edad media fue de 32,5 años, el 56,1% eran mujeres. El 36,5% de los pacientes eran homocigotos para Phe508del, el 40,4% heterocigoto y el 22,8% tenían otra mutación. El FEV1 medio fue de 72,2%. El 33,3% de los pacientes estaban colonizados por Pseudomonas aeruginosa (PA) sensibles y 7,0% por PA multirresistentes. Los costes totales anuales fueron de 623.981,3 euros (87,6% de costes de medicamentos y 12,4% de gastos médicos directos).
Los costes médicos, farmacéuticos y totales fueron mayores en el grupo de mutación Phe508del/Phe508del, que en Phe508del/otro y otros (p<0.05). La colonización microbiana aumentó los costes (p<0,05); la colonización por PA sensibles supuso costes más altos de fármacos y totales, de manera similar para PA multirresistentes, todas las diferencias estadísticamente significativas. Los costes médicos aumentaron con el nivel de gravedad de la función pulmonar (p<0,001), también los costes de medicamentos y totales, con la excepción de los pacientes más graves.
Conclusión: La CF es una enfermedad relativamente costosa para el sistema de salud. En nuestro estudio, los pacientes con mutación homocigota Phe508del, los valores bajos de FEV1 y la colonización tuvieron un costo más alto.
Palabras clave: Antibiotico, mutación CFTR, análisis de coste, fibrosis quística, FEV, Pseudomonas aeruginosa.
____
INTRODUCTION
Cystic fibrosis (CF) is an inherited, autosomal recessive disease, with high mortality, which has a significant economic impact on healthcare systems due to its complexity and the cost of its treatments, especially at present time with the introduction of new medications. In 2014, approximately 70,000 people worldwide had been diagnosed with CF1. The incidence differs among populations, it is remarkably more common in the white populations of Europe and North America than in the Asian and African populations, with variability within each country. In Europe, the prevalence is estimated to be 1-9 cases per 100,000 people2. However, given the unreported cases of this disease, this value could represent an underestimation3.
CF is produced by a mutation in the gene of the cystic fibrosis transmembrane conductance regulator (CFTR) The CFTR protein functions as a chloride channel, which helps maintain the proper balance of salt and water within a cell. Mutations in one or both copies of this gene cause chronic pulmonary, hepatic, and pancreatic dysfunctions that are responsible for most of the morbidity and mortality that ensues due to CF4.
Although this disease still has no cure, life expectancy has enhanced greatly in recent years. The median survival age is approximately 51 years for some patients that have CF5. This fact is related to a premature diagnosis of the disease; a comprehensive multidisciplinary care in specialized centers; an improvement in nutritional support; an active treatment of pulmonary and digestive complications; lung transplant; as well as new inhaled therapies and anti-infective treatments6. In addition, new therapeutic strategies, including CFTR modulating drugs, gene therapy, and mRNA repair are expected to further increase life expectancy1,3.
As mentioned before in this work, despite its low prevalence, CF may have a considerable impact on healthcare systems expenditures. For instance, an international retrospective prevalence-based study of adult patients with CF describes the annual direct cost per patient in Europe, with values between EUR 13,798.13 in Poland and EUR 42,381.80 in the United Kingdom7. Severity of CF disease based on FEV1 values and chronic Pseudomonas aeruginosa airway infection are related to higher direct costs8. Furthermore, due to the introduction of CFTR modulators (all of them orphan medicinal products), an increase in expenditures per patient is expected9.
The main aim of this work was to perform an economic assessment of direct medical costs (outpatient medical intervention and outpatient drug costs) of CF patients in an adult CF Unit at a third level hospital in Spain. The secondary objective was to evaluate the association between FEV1 values, chronic infection by main microorganisms, CFTR mutation, and direct medical costs.
METHODS
1. Literature review
We reviewed relevant literature on costs of CF from the last 15 years. The following databases were employed: MEDLINE, Web of Science, and EMBASE. As a mandatory requirement, the search words had to be part of the title or abstract. The search terms used were ‘cystic fibrosis’, ‘costs and costs analysis’, ‘economic burden’, ‘comparison of costs’, ‘costs of illness’, ‘health care costs’, and ‘social impact of cystic fibrosis’. Language was restricted to English. No other limitations were specified. A search on the references cited in the reviewed papers was also performed.
Inclusion criteria for publications in this review were as follows: 1) analysis of cost for adult population (>18 years of age); 2) average value of total CF treatment costs estimated in the study; and 3) cost analysis conducted between 2000 and 2017.
2. Study design
It was a retrospective observational study throughout the year 2017. The inclusion criteria were: 1) age above 18 years; 2) diagnosis of CF; 3) follow up by Department of Pneumology during 2017; and 4) medication collection from this hospital during 2017. Patients without complete annual monitoring were excluded. The study was approved by the Ethics Committee of the hospital.
3. Demographic and clinical variables
All patient data were collected from electronic history records and the outpatient electronic prescription system. The variables studied in this work bestowed demographic and clinical information. The demographic variables assessed were age, sex, and body mass index (BMI). Meanwhile, the studied clinical variables were mutation of CFTR; presence of mellitus diabetes (MD); pancreatic insufficiency; occurrence of massive haemoptysis; microbial and fungal colonization; forced vital capacity (FVC); FVC in one second (FEV1); FEV1/FVC relation; exacerbations with oral and intravenous antibiotics; number of outpatient pulmonology, digestive, endocrinology, and physiotherapist visits; number of spirometrys; number of glucose response curves; number of blood tests; number of sputum microscopy; number of ultrasound tests (fibroscan and digestive ultrasonography); number of radiology tests (chest radiograph and bone densitometry); and number of high resolution computerized axial tomography (HRCT), and abdominal computerized axial tomography (CT) scans. All of the medical variables considered were directly related to the disease.
BMI was calculated as weight [kg] divided by height squared [m2]. Weight and height were measured during medical consultation.
CFTR mutations were classified in Phe508del/Phe508del (homozygous for the deletion), Phe508del/other (heterozygous for the deletion) and other mutations.
Mellitus diabetes was defined as fasting blood glucose greater than 126 mg/dL and postprandial (at two hours) greater than 200 mg/dL.
The existence of pancreatic insufficiency was considered when the patient required the use of complement pancreatic enzymes with fecal elastase levels lower than 200 μg/g9.
Massive haemoptysis was defined as expectoration of blood with volume over 240 ml, following the guidelines of the Pulmonary Therapies Committee of the Cystic Fibrosis Foundation10.
Microbiologic colonization was considered as isolation of one or more microorganisms in 50% or more of sputum samples recollected in all medical visits during a year, and analysed by the Microbiology Department (according to Leeds criteria)11.
Forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) were estimated as part of the respiratory functional study by a spirometer. One spirometry was done in each medical consultation, taking as normal FVC and FEV1 values greater than 80% of the theoretical value. Results lower than 70% in the FEV1/FVC ratio were indicative of obstruction. Patients were stratified according to FEV1 values in dependence of lung function-related severity level, as follows: normal (FEV1 ≥81%), mild (80.9% to 61%), moderate (60.9% to 40%), and severe (≤39.9%)
Exacerbations in treated patients were grouped as mild/moderate (with oral antibiotics treatment) and severe (with parenteral antibiotics). The distinction between intravenous or inhaled administration was not considered.
4. Resource costs
Laboratory test prices were calculated based on hospital laboratory price lists. Outpatient visits and medical tests prices were calculated based on the price list for medical procedures at the hospital. Drug prices considered were laboratory-selling prices notified in Nomenclator (database with drug information about funding, prices and others). Since Ivacaftor, the drug that acts directly in the ion transport of defective cell-surface CFTR protein, was not commercialized in Spain during this study, its cost was assumed to be zero. To calculate the cost of the 7% saline solution, formulated at the Hospital Pharmacy, the price of the components utilized in its preparation was considered. Hereafter all amounts are specified in Euros.
5. Data analysis
Proportion of patients, means, standard deviation (SD), medians, interquartile range (IQR) of cost were calculated using SPSS Version 22.0 software. Depending on the type of data, parametrical or non-parametrical, t-student, ANOVA, or U-Mann Whitney of Kruskal Wallis models were employed. Multivariate regression analysis (Generalized linear Model) were utilized to identify the main cost drivers of total, drug, and medical costs. Results were considered statistically significant if the p-value was less than 0.05.
6. Compliance with ethics guidelines
The research was reviewed and approved by the Ethics Committee of La Princesa University Hospital. All procedures were in accordance with the Helsinki Declaration and its later amendments or comparable ethical standards. An informed written consent was not necessary because of the nature of this study.
RESULTS
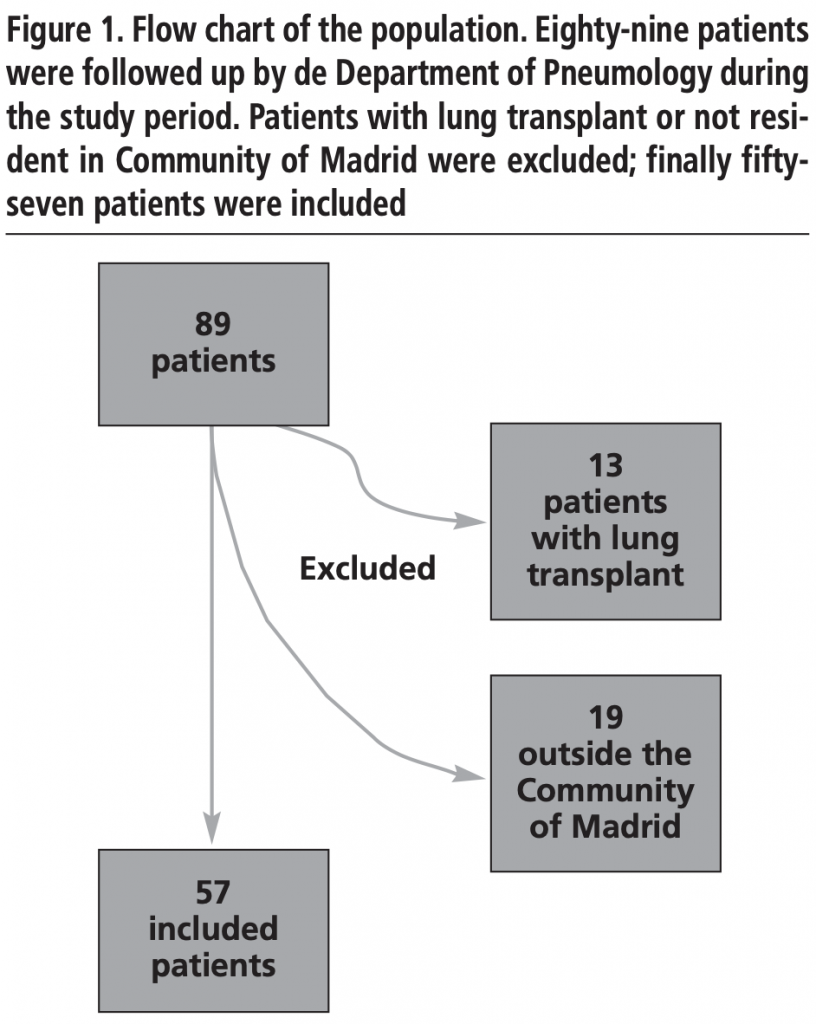
Eighty-nine CF patients were followed up by Department of Pneumology during 2017, and finally fifty-seven patients were included in the analysis. (Figure 1)
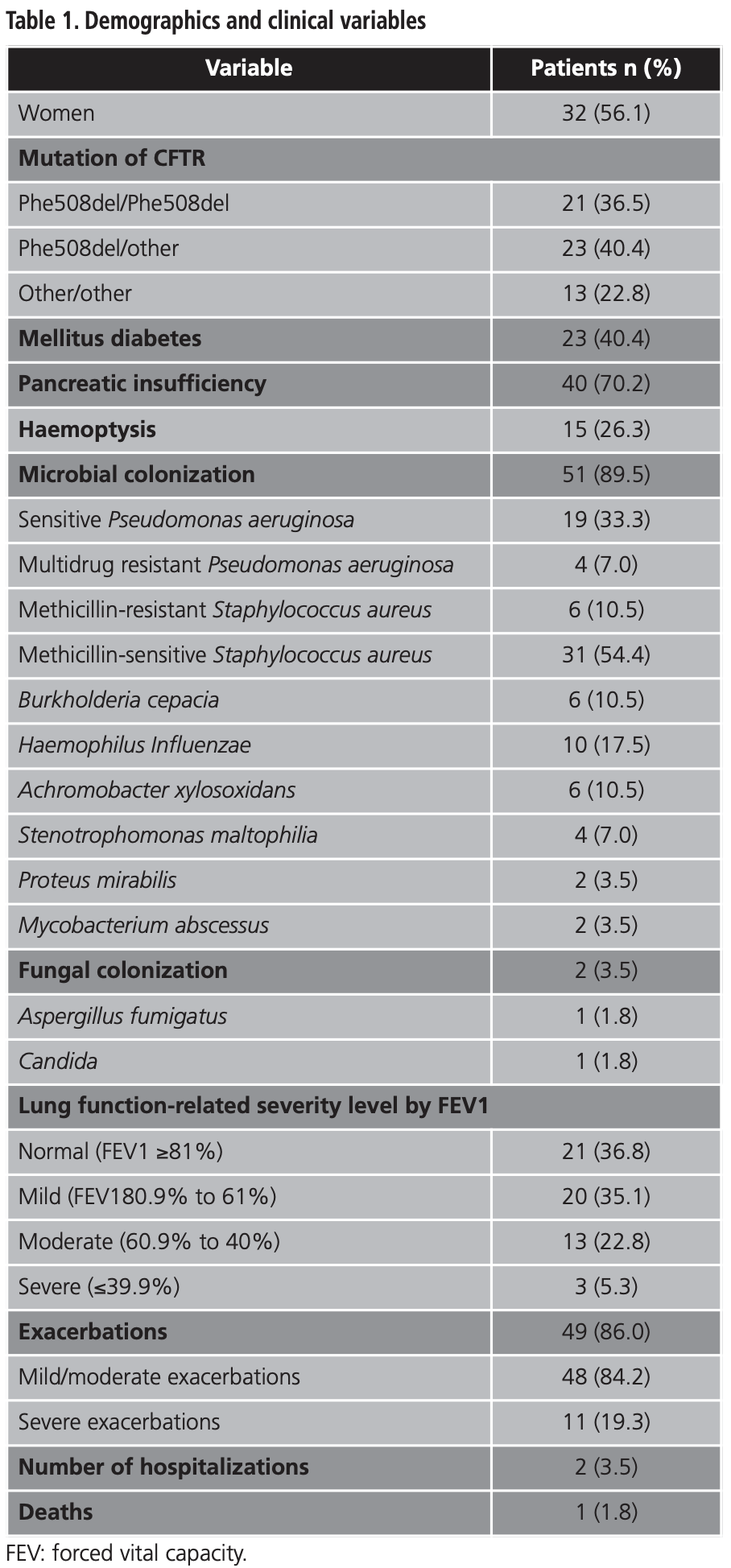
Demographic and clinical variables are presented in table 1. The mean (SD) age was 32.5 (9.2) years and 32 (56.1%) patients were female. The mean (SD) BMI was 22.7 Kg/m2 (3.1). The average (SD) FVC was 84.0% (19.6), FEV1 was 72.2 (21.2), and FEV1/FVC relation was 67.9 (12.3). The mean (SD) mild/moderate and severe exacerbations were 2.8 (2.3) and 3 (8.0), respectively.
The total costs per year were EUR 623,981.3, more than two thirds were attributed to drug costs related to medical costs (EUR 546,762.9 (87.6%) vs. EUR 77,218.3 (12.4 %)). The median total cost per patient was EUR 8,845.3, and ranged from 343.9 to 32,895.6. The mean (SD) medical cost was EUR 1,354.7 (513.9) and the median (IQR) drug cost was EUR 7,226.2 (5.5-30,868.0).
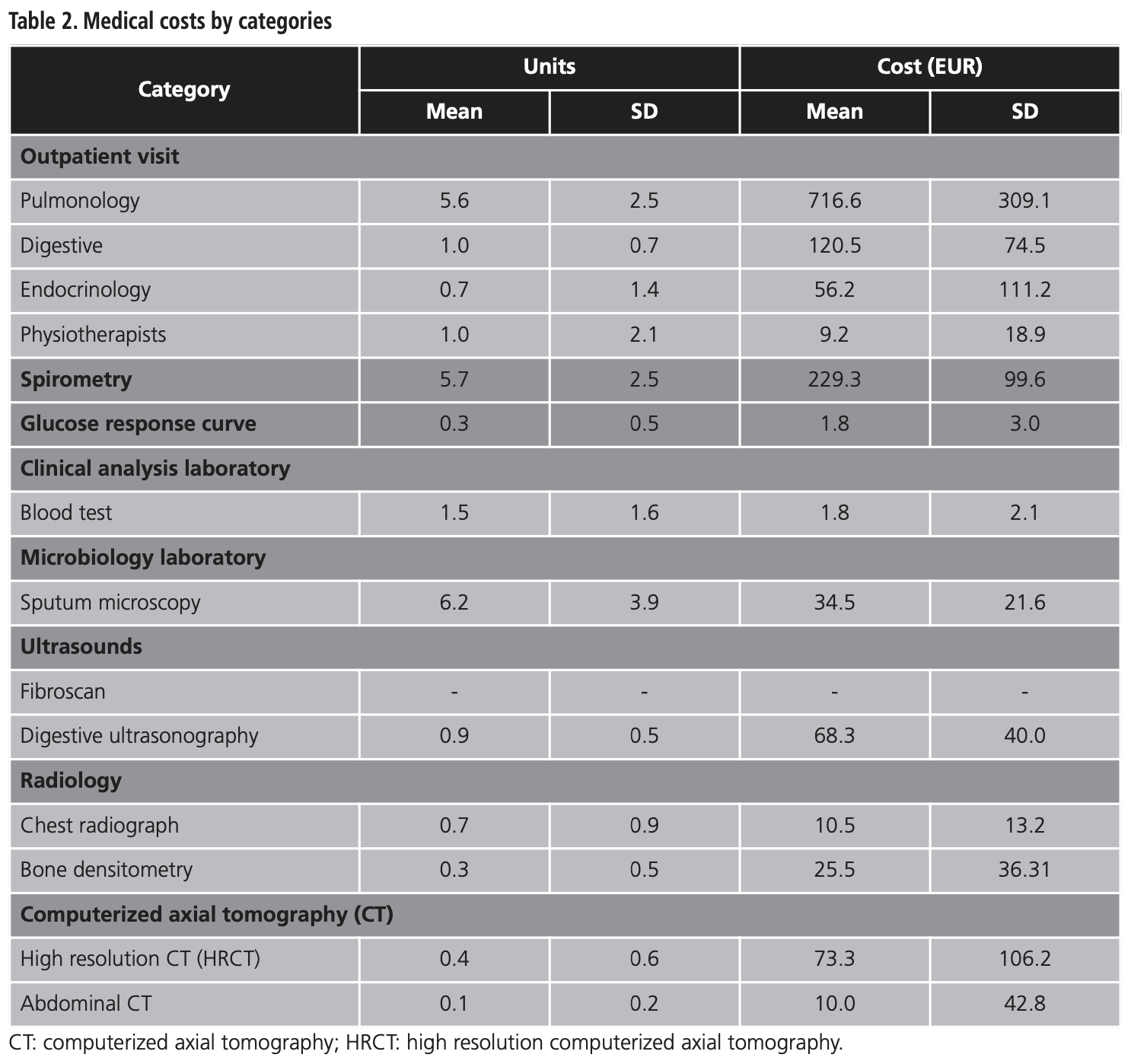
Medical costs by categories are shown in table 2. The main cost was the pulmonology visit, which corresponds to a mean (SD) cost of EUR 716.6 (309.1), followed by the spirometry mean cost (SD) of EUR 229.3 (99.6), and the digestive visit, with a mean cost (SD) of EUR 120.5 (74.5). Fibroscans were not performed to any patient during the study.
Drug groups with highest costs per year were Bramitob® (tobramycin) (EUR 58,326.2), Xolair® (omalizumab) (EUR 53,174.9), Promixin® (colistimethate) (EUR 47,559.4), and others parenteral or inhaled antibiotics (EUR 41,805.0).
The subgroup analysis is presented in table 3. Medical costs of females were higher than those of males, and the difference was statistically significant (EUR 1,525.1 vs. EUR 1,132.7, p=0.003), however drug costs and total costs were similar in both groups.
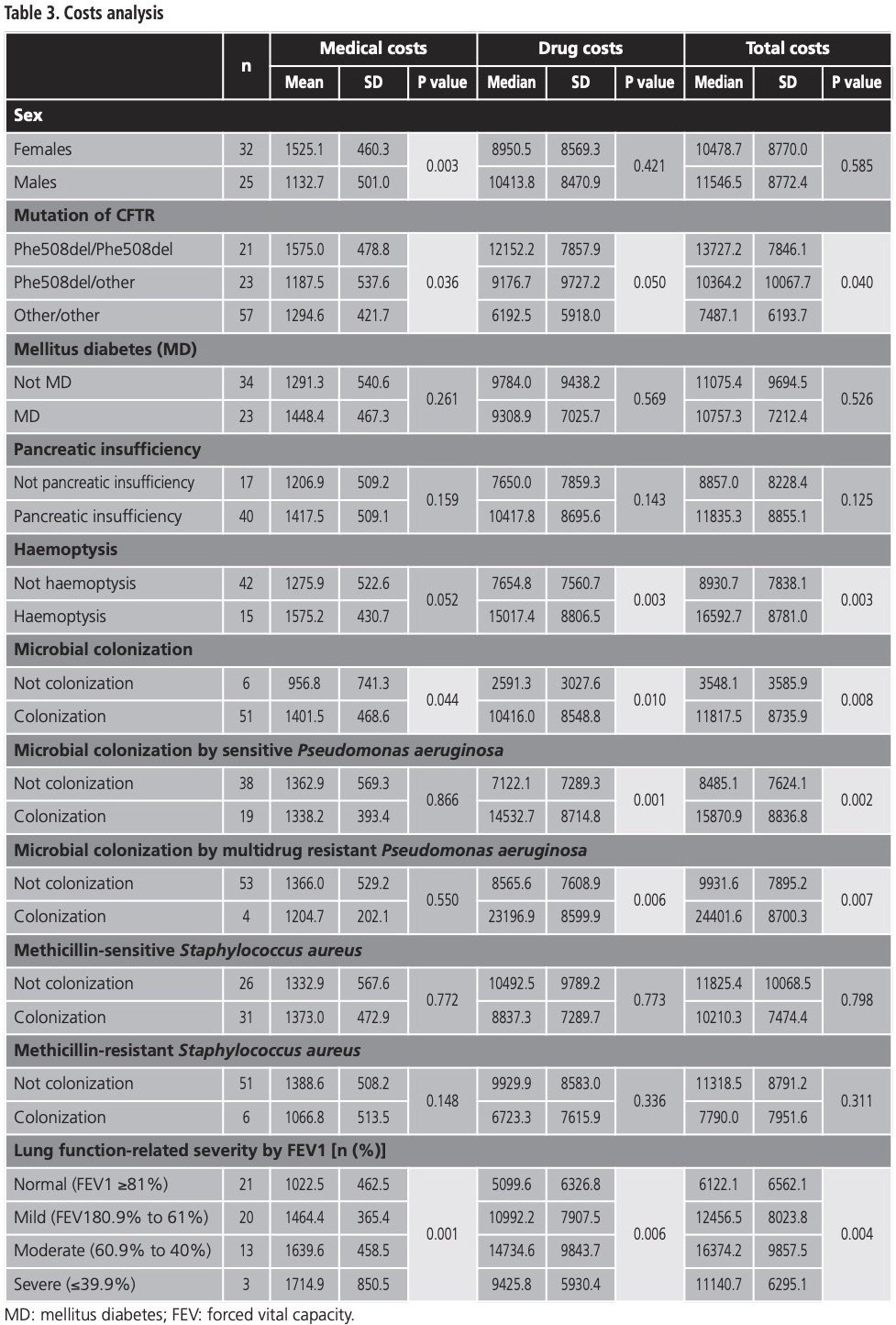
The ANOVA test of the mutation of CFTR groups shows that differences in medical costs were statistically significant, implying greater costs for the Phe508del/Phe508del mutation group (worst prognosis), as opposed to the Phe508del/other, and other/other mutation groups (EUR 1,575.0 vs. EUR 1,187.5 vs. 1,294.6, p=0.036); likewise the drug costs behaved similarly (EUR 12,152.2 vs. EUR 9,176.7 vs. EUR 6,192.5, p=0.050); and, therefore, total costs exhibited a comparable pattern (EUR 13,727.2 vs. EUR 10,364.2 vs. EUR 7,487.1, p=0.040).
Patients with haemoptysis correlated with higher drug- and total-costs, as opposed to those who did not exhibit haemoptysis (drug cost EUR 15,017.4 vs. EUR 7,654.8, p=0.003 and total cost EUR 16,592.7 vs. EUR 8,930.7, p=0.003, respectively). Some microbial colonizations sharply increased total costs (EUR 11,817.5 vs. EUR 3,548.1, p=0.008), as well as medical and drug costs, all differences were statistically significant. Patients colonized with sensitive Pseudomonas aeruginosa had statistically significant higher drug costs (EUR 14,532.7 vs. EUR 7,122.1, p=0.001), and total costs (EUR 15,870.9 vs. EUR 8,485.1, p=0.002). A similar scenario ensued with the multidrug resistant Pseudomonas aeruginosa aeruginosa classification (EUR 23,196.9 vs. EUR 7,608.9, p=0.006 and EUR 24,401.6 vs. EUR 9,931.6, p=0.007, respectively).
When it comes to lung function, medical costs increase with severity level: the difference between normal (EUR 1,022.5), mild (EUR 1,464.4), moderate (EUR 1,639.6), and severe (EUR 1,714.9) severity were statistically significant (p=0.001). Drug costs also increase among lung function severity, with the exception of severe patients, where expenditure was lower (EUR 5,099.6 vs. EUR 10,992.2 vs. EUR 14,734.6 vs. EUR 9,425.8, respectively, p=0.006). A similar situation occurs in total costs (EUR 6,122.1 vs. EUR 12,456.5 vs. EUR 16,374.2 vs. EUR 11,140.7, p=0.004).
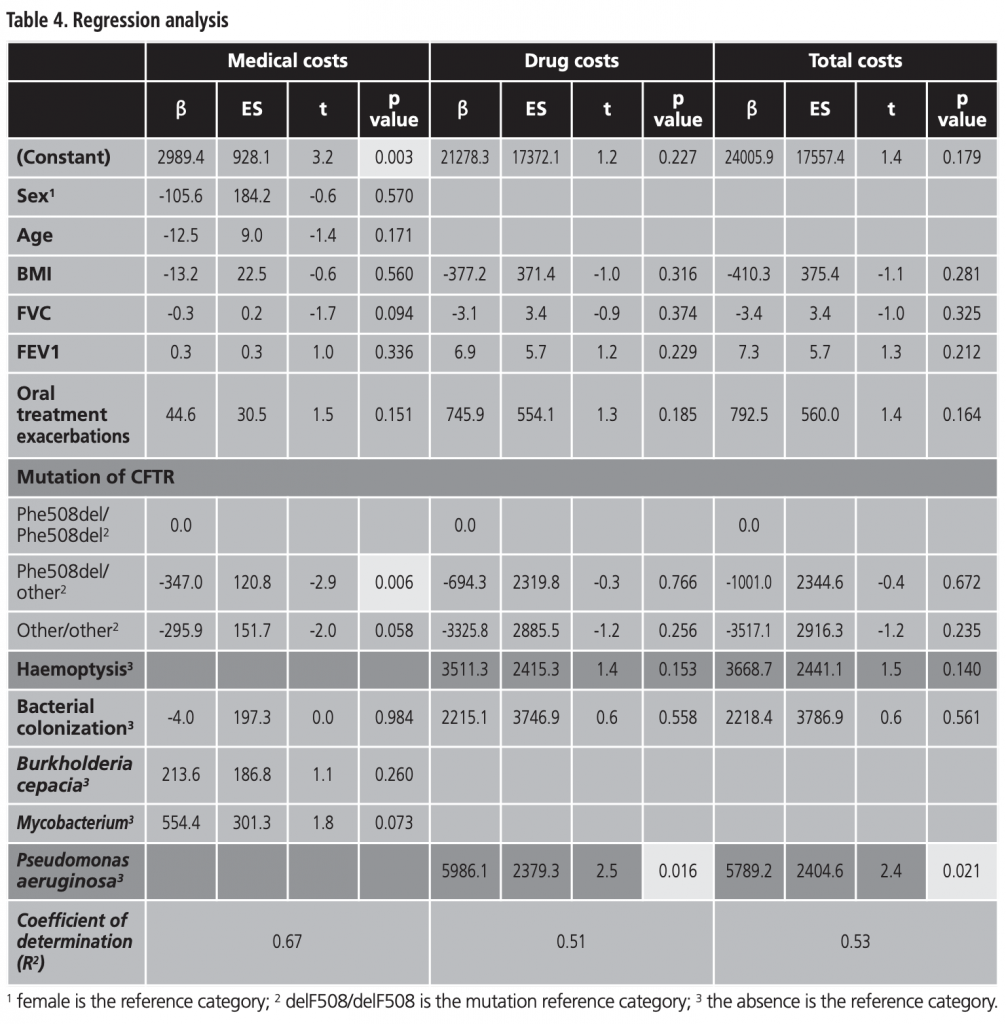
The multiple linear regression model for medical-, drug-, and total- costs (adjusted for sex, age, BMI, FVC, FEV1, mild/moderate exacerbations, mutation, haemoptysis and bacterial colonization) presents a good explanatory capacity (coefficient of determination=0.67; F=19.459, p<0.001). It included the variable “mutation” for medical costs (β coefficient=-347.0, p=0.006). Drug costs included Pseudomonas aeruginosa colonization (β coefficient=5,986.1, p=0.016) and total costs included Pseudomonas aeruginosa colonization also (β coefficient=5,789.2, p=0.016) (Table 4).
DISCUSSION
Considering rare diseases, CF represents a health problem with a significant social impact in first world countries13. The effect of CF on health, quality of life and the economic cost of this disease justify the attention given to the disease by both the society and its economic experts in medical care14.
To the best of our knowledge, this is the first attempt to estimate the care cost of patients with CF in Spain. Due to this fact, these results cannot be compared with other national reports. Although there are multiple studies from other countries, the cost calculation and regression models are often different in each study and therefore not comparable15. Taking this fact into consideration, we emphasize that our discussion is not a direct comparison. Instead it is a description of results presented against results from other countries.
The cost analysis was based on individual patient care data from a population of 57 CF patients who were seen systematically between January 2017 and December 2017 in a reference center for CF in Spain, La Princesa University Hospital in Madrid. According to a current regulation in Spain (Circular 8/1991, April 23), CF patients are followed in reference centers and all drugs must be prescribed and dispensed by the reference center. This system greatly favoured a reliable monitoring of drug utilization in our study.
In our work, solely outpatient costs were taken into consideration. We divided costs into two groups: 1. “Treatment costs” including costs associated with drugs and dietetic products; and 2. “Medical costs” including device prescriptions, medical tests, and medical consultations.
While previous studies have found that patients who have a mutation of homozygous Phe508del-CFTR are associated with high morbidity and mortality, our study was one of the first studies to estimate the effect of the genetic profile on the costs. We found that, on average, treating patients presenting homozygous Phe508del mutation costs twice as much than treating patients with other mutations (EUR 13,727 vs. EUR 7,487) (p<0.05). The Phe508del mutation leads to greatly reduced CFTR protein activity owing to impaired processing and trafficking of CFTR to cell surface, as well as impaired function of the small quantity of the protein to epithelial membranes4. Similar findings were previously reported by Jackson et al.6, Mlcoch et al.8, and Gu et al.16. Genetic profiles are relevant because CFTR mutation grouping is predictive of prognosis17, and the recent development of mutation-specific therapies to treat the underlying genetic defect have shown promising results18,19.
The results also demonstrate a direct correlation between chronic bacterial colonization and costs. Furthermore, medical costs were significantly higher with chronic colonization by Pseudomonas aeruginosa (EUR 14,532 vs. EUR 7,122), and treatment costs were significantly higher with colonization by Burkholderia cepacia (EUR 1,540 vs. EUR 1,332); both bacteria have been linked to clinical and functional impairment as well as an increase in the number of respiratory excretions4. The association between chronic bacterial colonization and costs has been well validated6,16,20-22.
As we pointed out previously in our discussion, although sometimes it is difficult to compare the results between studies because of the differences in the methodologies, almost all studies (including this work) found that FEV1 has a significant effect on the costs6,7,16,20,23,24. Nevertheless, severe lung function showed lower medical, drug and total costs than moderate lung function. This behavior could be due to the smaller number of patients included in this group.
In spite of not having a cure for this disease, expected lifespan has improved significantly recently, being the median survival age approximately 51 years for some patients with CF5. Due to this fact it is especially interesting to consider how age impacts the cost of this illness. Jackson et al.6, Gu et al.16, and Van Gool et al.12 found a positive effect of age on costs, being lowest in children, peaked in adults aged in their late twenties, and declining for older patients. This model is probably related to patients with better health, who require less medical intervention and therefore imply less costs, that are likely to live longer. Nevertheless, in our study we could not demonstrate this correlation.
CFTR modulator drugs are changing the FQ therapy. Ivacaftor (Kalydeco®) was financed in 2014 by EMA (PVL notified = EUR 18,000) for treatment of patients aged 6 years and older who have the G551D mutation. Meanwhile, lumacaftor/ivacaftor (Orkambi®) was approved in 2016 for treatment of patients aged 6 years and older who have the Phe508del mutation, although its funding and price is not currently established according to the Nomenclator database. Tezacaftor/ivacaftor (Symkevi®) has been demonstrated in clinical trials to improve the benefit-to-risk profile in patients with homozygous for Phe508del mutation25, and has been approved in January, 2019. Currently in our hospital, 1 patient is being treated with ivacaftor, 1 with lumacaftor/ivacaftor, and 2 with tezacaftor/ivacaftor, these last two combinations are part of an expanded access program.
The major limitation of the current study is that it was carried out in a single centre, enrolling a relatively limited sample size (n=57). Due to this limitation, these results cannot be projected to the entire population of CF patients in Spain, and therefore the data obtained have only informative value. Additionally, in our study we considered solely outpatient costs, so our results are not comparable with those of the commented studies that took into account inpatient and/or indirect costs.
At the moment, CF is a relatively costly disease for the Spanish healthcare system. In this study, we have shown that there are factors that contribute to increase significantly the cost of the disease. More specifically, we have shown that patients with the homozygous Phe508del mutation, decreased values of FEV1, and microbiological colonization will entail higher medical and drug costs.
We expect that this scenario will change during the upcoming years, when the CFTR drugs (lumacaftor/ivacaftor, tezacaftor/ivacaftor and other molecules in research and development) will finally be incorporated into routine clinical practice. A new economic analysis will be necessary to demonstrate the impact of such new CF treatments in healthcare system.
Conflict of interest: The authors declare that they have no conflict of interest.
BIBLIOGRAPHY
1. Schmidt M, et al. Strategies for new-born screening of cystic fibrosis: A systematic review of health economic evaluations. Journal of Cystic Fibrosis. 2018;17(3):306-315.
2. Orphanet: Cystic fibrosis. [Internet]. (cited 2019 Jan) Available from: http://www.orpha.net.
3. Stephenson AL, Stanojevic S, Sykes J, Burgel PR. The changing epidemiology and demography of cystic fibrosis. La Presse Medicale. 2017;46(6):87-95.
4. Brown SD, White R, Tobin P. Keep them breathing: cystic fibrosis pathophysiology, diagnosis, and treatment. Journal of the American Academy of Physician Assistants. 2017;30(5):23-27.
5. Stephenson AL, et al. Survival comparison of patients with cystic fibrosis in Canada and the United States: a population-based cohort study. Annals of Internal Medicine. 2017;166(8):537-546.
6. Jackson AD, et al. Estimating direct cost of cystic fibrosis care using Irish registry healthcare resource utilisation data, 2008-2012. PharmacoEconomics. 2017;35(10):1087-1101.
7. Kopciuch D, Zaprutko T, Paczkowska A, Nowakowska E. Cost of treatment of adult patients with cystic fibrosis in Poland and internationally. Public Health. 2017;148: 49-55.
8. Mlcoch T, et al. Cost-of-illness analysis and regression modeling in cystic fibrosis: a retrospective prevalence-based study. The European Journal of Health Economics. 2017;18(1):73-82.
9. Vadagam P, Kamal KM, Covvey JR, Giannetti V, Mukherjee K. Cost-effectiveness and budget impact of lumacaftor/ivacaftor in the treatment of cystic fibrosis. Journal of Managed Care & Specialty Pharmacy. 2018;24(10):987-997.
10. Domínguez-Muñoz JE, Hyeronimus C, Sauerbruch T, Malfertheiner P. Fecal elastase test: evaluation of a new noninvasive pancreatic function test. The American Journal of Gastroenterology. 1995;90:1834-1837.
11. Flume PA, et al. Cystic fibrosis pulmonary guidelines: pulmonary complications: haemoptysis and pneumothorax. American Journal of Respiratory Critical Care Medicine. 2010;182(3):298-306.
12. Lee TW, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. Journal of Cystic Fibrosis. 2003;2(1):29-34.
13. van Gool K, Norman R, Delatycki MB, Hall J, Massie J. Understanding the costs of care for cystic fibrosis: an analysis by age and health state. Value in health. 2013;16(2):345-355.
14. Angelis A, et al. Social and economic costs and health-related quality of life in non-institutionalised patients with cystic fibrosis in the United Kingdom. BMC Health Services Research. 2015;15:428.
15. Heimeshoff M, Hollmeyer H, Schreyögg J, Tiemann O, Staab D. Cost of illness of cystic fibrosis in Germany: Results from a large cystic fibrosis centre. PharmacoEconomics. 2012;30(9):763-777.
16. Gu Y, García-Pérez S, Massie J, van Gool K. Cost of care for cystic fibrosis: an investigation of cost determinants using national registry data. The European Journal of Health Economics. 2015;16(7):709-717.
17. McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest. 2006;130(5):1441-1447.
18. Davies JC, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. American Journal of Respiratory and Critical Care Medicine. 2013;187(11):1219-1225.
19. Bonnie W, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. New England Journal of Medicine. 2011;365(18):1663-1672.
20. Johnson JA, Connolly MA, Jacobs P, Montgomery M, Brown NE, Zuberbuhler P. Cost of care for individuals with cystic fibrosis: a regression approach to determining the impact of recombinant human DNase. Pharmacotherapy. 1999;19(10): 1159-1166.
21. Ouyang L, Grosse SD, Amendah DD, Schechter MS. Healthcare expenditures for privately insured people with cystic fibrosis. Pediatric Pulmonology. 2009; 44(10):989-996.
22. Eidt-Koch D, Wagner T, Mittendorf T, Graf von der Schulenburg JM. Outpatient medication costs of patients with cystic fibrosis in Germany. Applied Health Economics and Health Policy. 2010;8(2):111-118.
23. Colombo C, et al. Cost of Cystic Fibrosis: Analysis of treatment cost in a specialized center in Northern Italy. Advances in Therapy. 2013;30(2):165-175.
24. Donaldson SH, et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. American Journal of Respiratory Critical Care Medicine. 2018;197(2):214-224.
____