Rev. OFIL 2017, 27;3:249-255
Fecha de recepción: 06/01/2017 – Fecha de aceptación: 10/03/2017
Moya-Gil A1, Martínez-Gómez MA2, Climente-Martí M3, Merino-Sanjuán M4
1 Farmacéutica Especilista en Farmacia Hospitalaria. Servicio de Farmacia. Hospital Universitario Dr. Peset. Valencia (España)
2 Doctora en Químicas. Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunidad
Valenciana (FISABIO). Servicio de Farmacia. Hospital Universitario Dr. Peset. Valencia (España)
3 Jefe de Servicio de Farmacia Hospitalaria. Servicio de Farmacia. Hospital Universitario Dr. Peset. Valencia (España)
4 Catedrática. Departamento de Farmacia y Tecnología Farmacéutica. Facultad de Farmacia. Universidad de Valencia (España)
____
Correspondencia:
Ana Moya Gil
Hospital Universitario Dr. Peset
(Servicio de Farmacia)
Avda. Gaspar Aguilar, 90
46017 Valencia
Correo electrónico: amoyagil@gmail.com
____
Resumen
Objetivo: Valorar la estabilidad química y compatibilidad física de dexametasona.
Método: Estudio experimental de 15 días de duración. Productos: dexametasona fosfato sódico y NaCl 0,9% 50 ml (Viaflo®). Se analizaron 4 muestras por duplicado (0,10; 0,16; 0,38 y 0,89 mg/ml) con fotoprotección (PL) y en presencia de luz (L), refrigeradas (5±2ºC). Estabilidad química: se evaluó variación de concentración mediante cromatografía líquida de alta resolución (HPLC) calculando T90. Método cromatográfico fue válido para linealidad, precisión, exactitud, selectividad y repetibilidad, también se determinó límite de detección y cuantificación. Compatibilidad física se evaluó: a) cambio de color, turbidez y precipitación; b) pérdida de volumen; c) variación de pH.
Resultados: No se observaron cambios de color, turbidez, precipitación, ni pérdida de volumen. La variación de pH fue ≤2,7% excepto en muestra 1 (5,6%). El método cromatográfico resultó adecuado en linealidad (r2=0,9997), precisión intra-día e inter-día ≤10%, exactitud (≤8%), selectividad (ausencia de interferencias), límite de detección (0,020 mg/ml) y cuantificación (0,068 mg/ml). La estabilidad química de dexametasona refrigerada PL y L fue de 10 días para todas las concentraciones estudiadas, menos para la muestra de dexametasona refrigerada 0,10 mg/ml PL que fue de 11 días.
Conclusiones: El estudio amplía a 10 días la estabilidad química y compatibilidad física de dexametasona refrigerada a concentraciones 0,10, 0,16, 0,38 y 0,89 mg/ml utilizada como pauta antiemética del tratamiento antineoplásico. Esto permite la preparación anticipada, utilizándose los estudios de estabilidad como herramienta Lean garantizando la calidad en la preparación, mejorando la eficiencia en la Unidad Oncología Farmacéutica.
Palabras clave: Estabilidad, dexametasona, tratamiento antiemético, Lean.
____
Introducción
La farmacoterapia antineoplásica no solo incluye los medicamentos antineoplásicos, también tratamientos de soporte y sintomáticos que contribuyen a mejorar la supervivencia y calidad de vida. Estos pacientes son grupo de riesgo y es por ello que múltiples asociaciones y sociedades ciéntificas internacionales como National Institute of Health Clinical Center1, American Society of Health-System Pharmacists2, International Society of Oncology Pharmacists Practitioners3 y American Society of Clinical Oncology junto con Oncology Nursins Society4 han desarrollado estrategias en la prevención de errores de medicación en el paciente onco-hematológico. A nivel nacional, el Grupo Español para el desarrollo de la Farmacia Oncológica perteneciente a la Sociedad Española de Farmacia Hospitalaria, publicó recomendaciones para la prevención, entre ellas, la centralización de la preparación5,6.
El abordaje del tratamiento antineoplásico en nuestro centro sigue los estándares nacionales e internacionales para la seguridad del paciente onco-hematológico con una visión interdisciplinar7. En este sentido, se apostó por la preparación centralizada en el Servicio de Farmacia del tratamiento integral antineoplásico incluyendo la pauta antiemética, la fluidoterapia y otro tratamiento de soporte que se requiera, aportando valor al proceso, dispensando al Hospital de Día el tratamiento integral ready to use8.
Otra recomendación para minimizar los riesgos y mejorar la calidad de los servicios prestados es la disponibilidad de programas de Gestión de la Calidad Asistencial7. Fue en 2008 cuando la Unidad de Oncología Médica y la Unidad de Oncología Farmacéutica se certificaron por primera vez en el Sistema de Gestión de Calidad Norma ISO 9001:2008, acción consolidada hasta hoy. Además, a finales de 2012 se implantó la metodología Lean Seis Sigma (Lean: reducción de tiempos; Seis Sigma: reducción de variabilidad, precursora de errores) para la mejora continua de la microgestión del proceso farmacoterapéutico del paciente onco-hematológico. En ambos casos, el objetivo fundamental es mejorar para dar al cliente el mayor valor agregado, mediante una mejora continua y sistemática de calidad, costes, tiempos de respuestas, variedad y mayores niveles de satisfacción. Tras valorar la satisfacción del paciente onco-hematológico mediante una encuesta voluntaria y anónima (tasa de respuesta 68% (n=141), se concluyó que a pesar de que la satisfacción global era buena o muy buena (94%), el tiempo de espera para el tratamiento era el criterio peor valorado (27% insatisfecho o muy insatisfecho)9. La preparación del tratamiento antineoplásico integral es un proceso pull, es decir, un proceso cuya producción se activa por demanda tras la confirmación diaria del tratamiento de manera individual por paciente. En este sentido, se identificó la preparación del se identificó la preparación del tratamiento antineoplasico integral individual por paciente en la cabina de seguridad biológica como cuello de botella. La preparación anticipada de la pauta antiemética podría ser una oportunidad de mejora para la reducción de tiempos en la preparación.
Los corticoides están indicados en la profilaxis y tratamiento de las náuseas y los vómitos inducidos por citostáticos a dosis bajas: 10-20 mg de dexametasona fosfato (DEX) intravenoso o por vía oral antes de iniciar la quimioterapia, y posteriormente si fuese necesario10. El equipo interdisciplinar responsable del tratamiento antineoplásico de nuestro centro, sigue las recomendaciones del tratamiento antiemético según las Guidelines de la National Comprehensive Cancer Network. La pauta antiemética intravenosa pre-quimioterapia se selecciona según el riesgo emétogeno del tratamiento antineoplásico: DEX 8 mg asociado al bajo riesgo, DEX 8 mg más un antagonista selectivo del receptor 5HT3 (ondasetrón 8 mg o granisetron 3 mg) asociado al riesgo intermedio y DEX 8 mg junto con ondasetrón 8 mg o granisetron 3 mg más un antagonista selectivo de alta afinidad por receptores de sustancia P neurocinina-1 (Fosaprepitant 150 mg) para alto riesgo11.
Tras una revisión bibliográfica sobre la estabilidad físico-química de DEX 8 mg en 50 ml de cloruro sódico 0,9% (NaCl 0,9%) en un envase de poliolefinas, no se encontraron resultados favorables para su preparación anticipada y conservación refrigerada (requisito de conservación para preservar estabilidad microbiológica)12. Según ficha técnica, las soluciones inyectables de DEX-Fortecortin® son compatibles con NaCl 0,9% y tienen que ser usadas en un plazo de 24 horas10. Otras fuentes bibliográficas: a) estabilidad de 91días en vídrio disuelto en agua para inyección con bacteriostático a una concentración de 10 mg/ml en presencia de luz (L) a temperatura ambiente o refrigerada; b) estabilidad de 22 días en un envase de polipropileno disuelto en suero fisiológico a dos concentracionesde 0,1 y 10 mg/ml, conservada a temperatura ambiente sin especificar condiciones de protección de luz; c) estabilidad de 55 días en polipropileno disuelto en agua para inyección con bacteriostático a una concentración de 10 mg/ml en L a temperatura ambiente o nevera; d) estabilidad de 28 días disuelto en suero fisiológico con bacteriostático a concentraciones desde 1 a 4 mg/ml en L a temperatura ambiente sin especifiar tipo de envase13.
El objetivo de este estudio ha sido valorar la estabilidad química y la compatibilidad física de DEX a diferentes concentraciones (0,1-0,9 mg/ml) en NaCl 0,9% en envase de poliolefinas conservada en nevera con y sin fotoprotección para su preparación anticipada.
MÉTODO
Estudio experimental en el que se tomaron muestras de las diferentes soluciones a distintos tiempos durante 15 días. Los tiempos de muestreo fueron: 0, 12, 24, 48, 72, 97, 122, 145, 167, 191, 212, 238, 262, 310 y 358 horas. A cada tiempo se evaluó la estabilidad química y la compatibilidad física siguiendo las directrices de la Farmacopea y las recomendaciones del Comité Internacional de Armonización (ICH)14-16.
Los productos de ensayo utilizados fueron: dexametasona fosfato sódico (Fortecortín®) 40 mg/5 ml inyectable, Laboratorio Merck S.L.; NaCl 0,9% volumen 10 ml, Laboratorio B. Braun Medical; NaCl 0,9% volumen 50 ml Viaflo® solución para perfusión, Laboratorio Baxter.
Los reactivos y material de laboratorio empleados fueron: Acetonitrilo (ACN), J.T.:Baker; ácido ortofosfórico (H3PO4) 85% (Fluka); agua estéril para irrigación volumen 1 litro (Baxter); jeringas estériles de polipropileno 6 ml (Terumo); agujas estériles 21G1/2 Microlance (Becton Dickinson); bolsas fotoprotectoras (Diffuplast); cámara frigorífica a (5±2)ºC (Vizuete S.L., España); columna analítica Kromasil C8 (4,6×250 mm, 5 µm) (Análisis Vínicos S.L., España); viales ámbar HPLC de 1,5 ml (Análisis Vínicos S.L., España); insertos para vial HPLC de 1,5 ml (Análisis Vínicos S.L., España).
Se utilizaron los siguientes instrumentos analíticos: Balanza analítica (GF-200, A&D Instruments Ltd, UK); pH-metro con electrodo de vidrio (Jenway, UK); Cromatógrafo líquido de alta resolución con detección ultravioleta-visible (HPLC; Agilent technologies, USA).
Procedimiento experimental
A) Preparación de las muestras
Muestra 1 y 2: concentración final 0,10 mg/ml (0,63 ml de Fortecortín® 40 mg/5 ml en 50 ml de NaCl 0,9% Viaflo®).
Muestra 3 y 4: concentración final 0,16 mg/ml (1,00 ml de Fortecortín® 40 mg/5 ml en 50 ml de NaCl 0,9% Viaflo®).
Muestra 5 y 6: concentración final 0,38 mg/ml (2,50 ml de Fortecortín® 40 mg/5 ml en 50 ml de NaCl 0,9% Viaflo®).
Muestra 7 y 8: concentración final 0,89 mg/ml (6,25 ml de Fortecortín® 40 mg/5 ml en 50 ml de NaCl 0,9% Viaflo®).
B) Condiciones de conservación del estudio
Todas las muestras se conservaron refrigeradas a una temperatura de (2±8)ºC. Además, las muestras 1, 3, 5 y 7 fueron protegidas de la luz (PL) mediante una bolsa fotoprotectora (presencia de luz: L).
C) Estudio de estabilidad química
Se evaluó la estabilidad química de DEX a diferentes concentraciones (0,10; 0,16; 0,38 y 0,89 mg/ml) en NaCl 0,9% mediante control analítico por HPLC. El parámetro T90 fue el criterio de estabilidad química: tiempo después del cual la concentración del fármaco remanente es inferior al 90% (tomando como referencia una desviación de 10% como control de calidad en cuanto a la seguridad y la eficacia de la preparación de las formulaciones magistrales como indica la Farmacopea)17. La concentración de DEX se expresó como porcentaje de variación con respecto a la concentración a tiempo 0. Se tomó, a diferentes tiempos y durante 15 días, una alícuota de 1 ml de cada muestra, previa homogeneización mediante 5 ciclos de doble inversión. Cada alícuota se analizó por duplicado.
Previo al inicio de cada sesión de trabajo, se acondicionó el sistema cromatográfico durante un mínimo de 30 minutos con fase móvil 25% acetonitrilo-75% H3PO4 a flujo 0,8 ml/min. Una vez estabilizado el sistema, se realizó análisis cromatográfico en las siguientes condiciones: volumen inyección: 20 µl; temperatura: 20ºC; flujo: 0,8 ml/min; fase móvil: t=0 minutos 25% ACN-75% H3PO4, t=7 y, t=14 minutos 50% ACN-50% H3PO4; λ: 241 nm. En estas condiciones, el tiempo de retención de DEX fue 8,7 minutos. De cada cromatograma se midió el área de pico de DEX. Para relacionar el área de pico con concentración, se construyó una recta de calibrado (área vs. concentración) a partir de disoluciones patrón de DEX de concentración conocida y creciente en 1 ml de NaCl 0,9%: Patrón 1: DEX 0,03 mg/ml; Patrón 2: DEX 0,10 mg/ml; Patrón 3: DEX 0,20 mg/ml; Patrón 4: DEX 0,40 mg/ml; Patrón 5: DEX 0,70 mg/ml; Patrón 6: DEX 1,00 mg/ml; Patrón 7: DEX 2,00 mg/ml. Cada disolución patrón se preparó por triplicado de manera independiente.
El método cromatográfico desarrollado fue validado en términos de linealidad, precisión, exactitud, selectividad y repetibilidad y se determinó el límite de detección y cuantificación, de acuerdo con las pautas de ICH18,19, analizándose las disoluciones patrón por triplicado durante 3 días consecutivos:
1- Linealidad: proporcionalidad entre área de pico de DEX (y) y concentración del mismo de las disoluciones patrón (x). y = m*x+b. Se tomó como criterio de aceptación que el coeficiente de correlación fuera ≥0,98. Se construyeron las correspondientes rectas de calibrado, se compararon los valores de las pendientes, los valores de las ordenadas en el origen con cero y los coeficientes de correlación con 119.
2- Precisión intra-día: se expresó como coeficiente de variación (CV) de la concentración de fármaco y se adoptó como criterio de aceptación que el CV sea ≤10±2,5%19.
3- Precisión inter-día: se expresó como CV de la concentración de fármaco y se adoptó como criterio que el CV sea ≤10±2,5%19.
4- Exactitud: se expresó como diferencia entre la concentración determinada y la teórica en tanto por cien y se adoptó como criterio de aceptación que la exactitud sea de 15%19 (X = concentración de fármaco determinada experimentalmente; CT = concentración de fármaco teórica para cada patrón).

5- Selectividad: se evaluó la posible interferencia de los excipientes presentes en las muestras al tiempo de retención de DEX.
6- Repetibilidad: se realizaron 10 análisis de cada una de las muestras 1, 3, 5 y 7. Se calculó como la desviación estándar (DS) de las medidas para cada muestra.
7- Límite de detección: concentración mínima de DEX presente en la muestra que puede ser detectable, pero no necesariamente cuantificable, en las condiciones experimentales de trabajo. Se calculó como 3*S/b, siendo S la DS de los residuales y b la pendiente de la recta de calibrado.
8- Límite de cuantificación: concentración mínima de DEX presente en la muestra que puede ser determinada con una precisión y exactitud aceptable en las condiciones experimentales de trabajo (<20%). Se calculó como 10*S/b.
D) Estudio de compatibilidad física
Se evaluó la compatibilidad física de DEX a diferentes concentraciones (0,10; 0,16; 0,38 y 0,89 mg/ml) en NaCl 0,9%, a diferentes tiempos y durante 15 días. Se tomó una alícuota de 5 ml de cada mezcla en estudio, previa homogeneización mediante 5 ciclos de doble inversión. Para cada alícuota se evaluaron los siguientes parámetros:
1- Aspecto: se controlaron variaciones en el color, formación de gas y aparición de precipitado mediante inspección visual: a) observación visual directa sobre fondo negro y blanco; b) comparación de las muestras con blancos de NaCl 0,9%.
2- Medida del pH: para cada una de las muestras preparadas se determinó el pH con un pH-metro con electrodo de vidrio.
3- Pérdida de volumen: el control de la variación de volumen por posible evaporación se realizó mediante gravimetría, pesando cada muestra antes y después de tomar la alícuota en cada tiempo de muestreo en una balanza analítica.
RESULTADOS
A) Estudio de estabilidad química
Validación del método analítico
El método cromatográfico para la determinación de DEX resultó adecuado en términos de linealidad, precisión, exactitud, selectividad y repetibilidad.
1- Linealidad: y = 16600,1·x, con un coeficiente de correlación = 0,9997 y ordenada en el origen, no estadísticamente significativa.
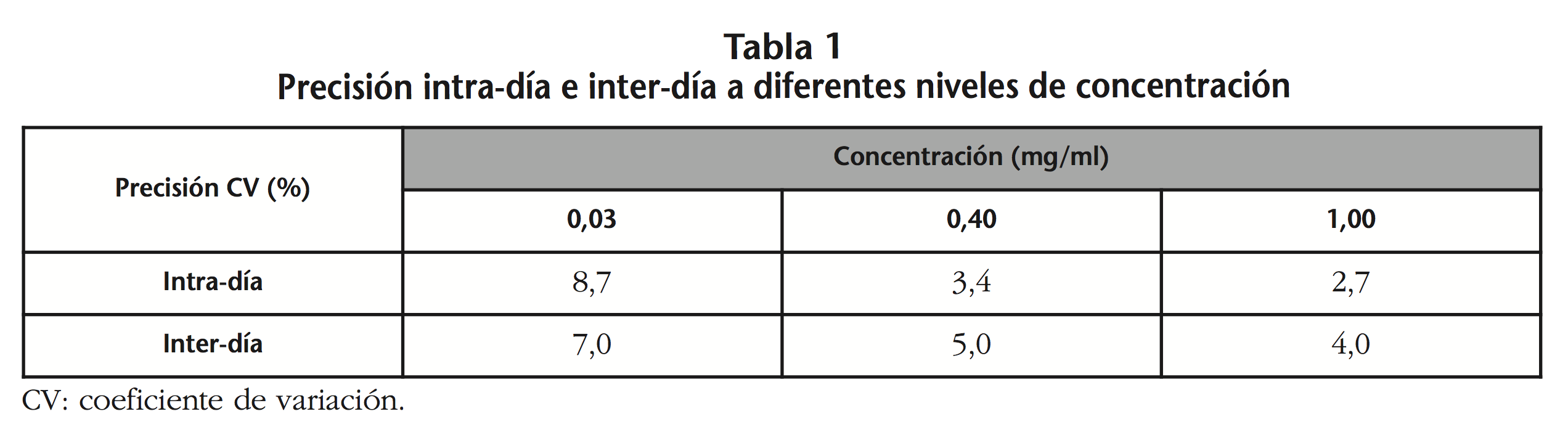
2- Precisión intra-día e inter-día: para todas las concentraciones el CV fue <10%. La tabla 1 muestra los valores de CV intra-día e inter-día para cada nivel de concentración ensayado.
3- Exactitud: los valores fueron 8,0%, 0,9% y 1,8%, para las concentraciones 0,03, 0,40 y 1,00 mg/ml, respectivamente.
4- Selectividad: no se observaron interferencias de los excipientes presentes en las muestras al tiempo de retención de DEX.
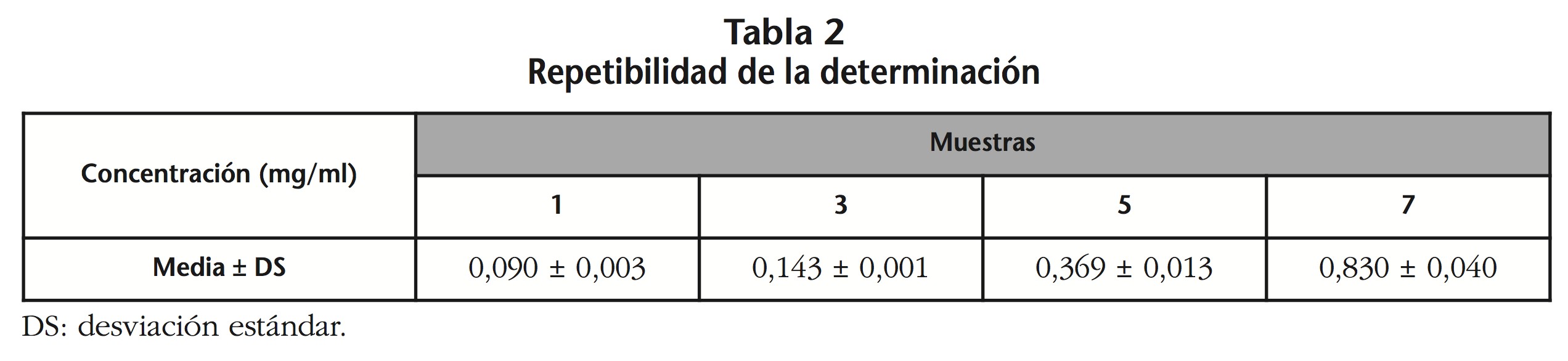
5- Repetibilidad: la tabla 2 muestra el valor medio y la DS de los 10 análisis efectuados de las muestras 1, 3, 5 y 7, siendo en todos los casos <1%.
6- Límite de detección y límite de cuantificación: fue 0,020 y 0,068 mg/mL, respectivamente.
Variación de la concentración de dexametasona con el tiempo
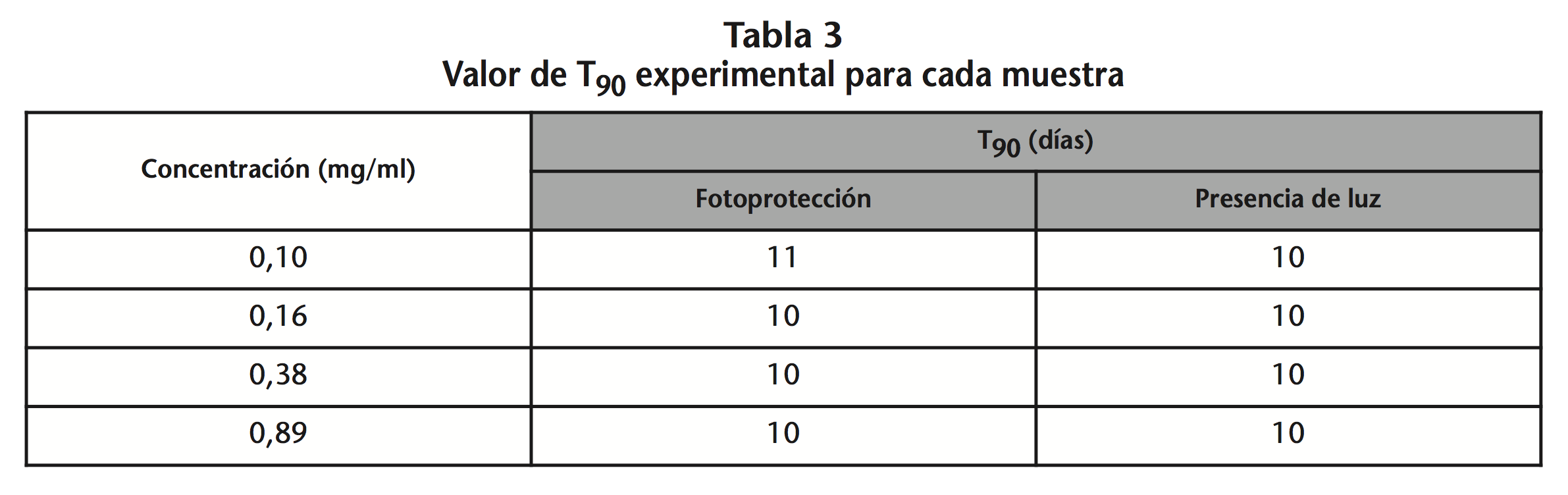
Al final del estudio, todas las muestras alcanzaron una concentración remanente de DEX en torno al 70%. Los pares de datos concentración-tiempo no se ajustaron en ningún caso a una cinética de orden 0 ni 1 y el valor de T90 correspondió al valor experimental medido. Los valores de T90 para todas las muestras fueron 10 días (240 h) tanto PL como L, menos para las muestras 0,10 mg/ml PL que fueron 11 días (264 h) (Tabla 3). En la figura 1 se representa la variación de la concentración de DEX con el tiempo para cada una de las soluciones.
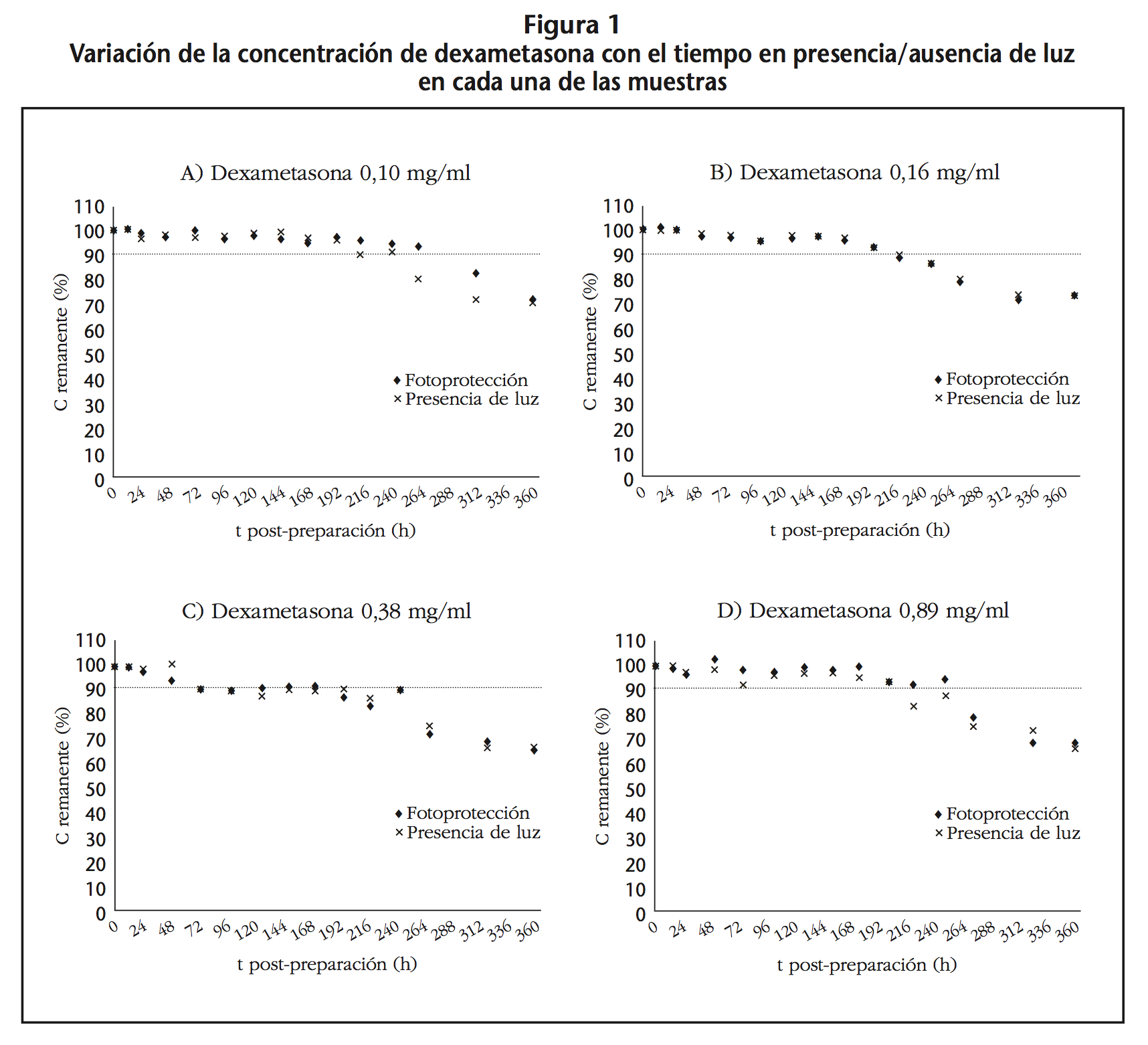
B) Estudio de compatibilidad física
No se observaron cambios de color, turbidez, precipitación, ni pérdida de volumen en ninguna de las muestras estudiadas durante el tiempo de estudio. Al final del estudio, la variación de pH fue ≤2,7% en todas las muestras excepto en la muestra 1, que fue del 5,6%.
DISCUSIÓN
El farmacéutico se encuentra en una situación óptima para identificar las oportunidades de mejora en la farmacoterapia de los pacientes y aportar valor a los resultados finales de la asistencia santiaria. Así pues, las prácticas de estudios de estabilidad, son ensayos necesarios para adaptarse a los principios guiados por las Organizaciones, mejorando los procesos farmacoterapéuticos y completando la información que facilita la industria farmacéutica garantizando la calidad del producto y la seguridad del paciente20.
La elaboración de mezclas intravenosas requiere de un control de calidad de la preparación, depende del uso correcto de los componentes, los cálculos realizados, la exactitud y la precisión del pesaje y el volumen, desde el respeto de los procedimientos y condiciones de funcionamiento adecuados, de modo que los preparados deben ser garantizados17.
Este estudio amplió a 10 días la información disponible sobre la estabilidad química y compatibilidad física de DEX 8 mg en 50 ml de NaCl 0,9% en envase de poliolefinas para su preparación anticipada y conservación refrigerada, que se adaptará a la práctica asistencial habitual. Sin embargo, las soluciones de DEX 20 mg en 50 ml de NaCl 0,9%, no fueron candidatas para su preparación antipada por su baja frecuencia de utilización10,13.
No obstante, para la preparación anticipada de mezclas intravenosas, no solo ha de considerarse la estabilidad química y compatibilidad física, otro criterio a tener en cuenta es la estabilidad microbiológica dependiendo de diferentes criterios como son las instalaciones utilizadas para la preparación y la propia preparación según indica la Chapter 797- Pharmaceutical compounding: sterile preparations. En este sentido, considerando un riesgo medio en cuanto a la contaminación de este tipo de preparaciones según instalaciones, procedimientos utilizados y otros criterios en Chapter 797, establecería una estabilidad microbiológica de 30 horas a temperatura ambiente y 9 días refrigeradas12. Sin embargo la estabilidad final, será la más restrictiva según la estabilidad físico-química o estabilidad microbiológica.
En este sentido, estudios como éste, garantizan la calidad y seguridad en el proceso farmacoterapéutico de antineoplásicos centrado en el paciente7. El diseño de este estudio ha permitido ampliar las condiciones adecuadas de preparación y conservación de DEX en su indicación como pauta antiemética a bajas dosis para su uso en la práctica clínica mejorando la calidad y seguridad del paciente onco-hematológico. Así pues, con la revisión bibliográfica y con otros estudios como éste, han permitido preparar de manera centralizada y anticipada por parte del Servicio de Farmacia5,6 las pautas antieméticas ready to use8 para el riesgo bajo (dexametasona 8 mg/NaCl 0,9% 50 ml), riesgo medio (dexametasona 8 mg/ondasetron 8 mg/NaCl 0,9% 50 ml o dexametasona 8 mg/granisetron 3 mg/NaCl 0,9% 50 ml)21,22 y riesgo alto (dexametasona 8 mg/ondasetron 8 mg/fosaprepitant 150 mg/NaCl 0,9% 100 ml o dexametasona 8 mg/granisetron 3 mg/fosaprepitant 150 mg/NaCl 0,9% 100 ml)23. Sin embargo, para el riesgo bajo-intermedio de emésis con dexametasona 20-40 mg/NaCl 0,9% 50 ml, no siendo candidata para su preparación anticipada por una incidencia <4,5%.
La preparación anticipada de las pautas antieméticas para disponer de ellas just in time y con una planificación mediante tarjetas kanban (según consumos, estabilidad química, compatibilidad física y estabilidad microbiológica), ha supuesto una reducción de 27 minutos en el tiempo de preparación del tratamiento antineoplásico integral con un promedio de 3.097 pacientes-día atendidos trimestralmente según datos disponibles en los indicadores del Sistema de Gestión de Calidad implantado (datos no publicados). Medidas como esta en la microogestión Lean Manufacturing (disminuir tiempos), siguiendo el Espíritu Kaizen maximiza el valor de la organización mediante la consecución de mayor satisfacción del cliente, el coste, la calidad y la velocidad del proceso obteniéndose resultados de efectividad y eficiencia24. Desde el año 2000, existe una tendencia emergente en el ámbito sanitario de la implantación de esta metodología, para la mejora de los procesos y la satisfacción de los pacientes, alguna de estas experiencias han reportado beneficios de 100.000€25,26. También existen experiencias en la Farmacia Hospitalaria donde la implantación de Lean Seis Sigma disminuyeron en un 50% los errores27,28.
CONCLUSIONES
El estudio amplía a 10 días la estabilidad química y compatibilidad física de dexametasona refrigerada a concentraciones 0,10, 0,16, 0,38 y 0,89 mg/ml para la indicación de pauta antiemética del tratamiento antineoplásico. Esto permite la preparación anticipada, utilizándose los estudios de estabilidad como herramienta Lean garantizando la calidad en la preparación mejorando la eficiencia en la Unidad Oncología Farmacéutica.
Conflicto de intereses: Los autores declaran no tener conflicto de intereses.
Bibliografía
1. Goldspiel BR, De Christoforo R, Daniels CE. A continuous-improvement approach for reducing the number of chemotherapy-related medication errors. Am J Health-Syst Pharm. 2000;15;Suppl 4:S4-9.
2. ASHP guidelines on preventing medication errors with antineoplasic agents. Am J Health-Syst Pharm. 2002; 59:1648-68.
3. Estándares de práctica para el manejo seguro de citotóxicos de la International Society of Oncology Pharmacists Practitioners (ISOPP). J Oncol Pharm Practice. 2007;13 supl 1:1-81.
4. American Society of Clinical Oncology/Oncology Nursing Society Chemotherapy Administration Safety Standards. ASCO/ONS Standards for safe chemotherapy administration [Internet]. 2009. [acutalizado 2013; citado 4 Dic 2016]. Disponible en: https://www. ons.org/practice-resources/clinical-practice/ascoons-chemotherapy-administration-safety-standards.
5. Grupo Español para el desarrollo de la Farmacia Oncológica (GEDEFO). Documento de consenso para la prevención para la utilización de medicamentos en la modalidad de uso compasivo en la terapéutica onco/hematológica. [Internet]. Mayo 2002.[actualizado 2002; citado 4 Dic 2016 ] Disponible en: http://gruposdetrabajo.sefh.es/gedefo/images/stories/documentos/Web_Doc_con_Compasivo.pdf.
6. Grupo Español para el desarrollo de la Farmacia Oncológica (GEDEFO). Documento consenso. Antineoplásicos orales. [Internet]. Octubre 2009. [actualizado Oct 2009; citado 4 Dic 2016] Disponible en: http://www. sefh.es/gedefo/documentos/consenso_gedefo_antineoplasicos_orales.pdf.
7. Jiménez Torres NV, Albert Martí A Almenar Cubells D, Vandenbroucke. La seguridad del paciente oncológico. Estándares internacionales para el manejo de citotóxicos. [Internet]. Real Academia Nacional de Farmacia. 12 de junio de 2008. [actualizado 2008; citado 3 Dic 2016]. Disponible en: http://www.analesranf.com/ index.php/funda/article/download/951/939.
8. Andrés Navarro N, Carreras Soler MJ, Cliemente Martí M, Moya Gil A, et al. Guía para la mejora de la seguridad del proceso farmacoterapéutico antineoplásico en pacientes onco-hematológicos. 1ª ed. Madrid: Sociedad Española de Farmacia Hospitalaria (SEFH); 2015.
9. Moya Gil A, Hernández Griso M, Climente Martí M, Porta Oltra B, et al. Satisfacción de los pacientes onco-hematológicos atendidos en Hospital de Día. [Internet]. Calidad en la era de las tacnologías de la información. Libro de Comunicaciones XXXI Congreso Nacional de la Sociedad Española de Calidad Asistencial y I Congreso de la Sociedad Valenciana de Calidad Asistencial. Sociedad Española de Calidad Asistencial. 1ªed. Valencia; octubre 2013 [actualizado 2013; citado 3 Dic 2016]. Disponible en: http://calidadasistencial. es/congresos/2013/galeria-de-imagenes/LIBRO_COMUNICACIONES_ORALES_DEFINITIVO.pdf.
10. Monografía Fortecortin® 40mg, 5ml. Merck, s.l. [Internet]. Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Centro de Información online de Medicamentos de la AEMPS; Ene 1987 [actualizado Sept 2016; citado 4 Dic 2016]. Disponible en: https://www.aemps.gob.es/cima/pdfs/es/ft/57036/FT_57036.pdf.
11. Antiemesis. NCCN Clinical Practice Guidelines in Oncology. [Internet]. Version 2.2016. National Comprehnsive Cancer Network; 2015 [acatualizado Feb 2016; citado 4 Dic 2016]. Disponible en: https://www. nccn.org/professionals/physician_gls/f_guidelines.asp.
12. Chapter 797. Pharmaceutical compounding: sterile preparations. [Internet]. USP31–NF26. The United States Pharmacopeia (USP). The ASHP Discussion Guide on USP Chapter for compounding: sterile preparation. June 2008. [actualizado Dic 2015; citado 11 Dic 2016]. Disponible en: http://www.ashp.org/doclibrary/policy/compounding/discguide797-2008.pdf.
13. Dexamethasone sodium phosphate. Stability in solution: Dexamethasone sodium phosphate. [Internet]. Stabilis: Stability and compatibility of drugs; September 2015 [actualizado Sept 2015; citado 3 Dic 2016]. Disponible en: http://www.stabilis.org/Monographie. php?IdMolecule=35&IdOnglet=StabSol#InfosSupp.
14. Guideance for Industry. Patient-Reported Outcome Measures: Uses in Medical Product Development to Support Labeling Claims. [Internet] US Department of Health and Human Services FDA Center forDrugEvaluation and Research; US Department of Health and Human Services FDA Center forBiologicsEvaluation and Research; US Department of Health and Human Services FDA Center forDevices and RadiologicalHealth; 2009 [actualizado Dic 2009; citado 8 Dic 2016]. Disponible en: http://www.fda.gov/downloads/Drugs/ Guidances/UCM193282.pdf.
15. ICH Harmonised Tripartite Guideline. Stability Testing of New Drug Substance and Products Q1A(R2) [Internet] International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2003 [actualizado 2003; citado 8 Dic 2016]. Disponible en: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2__Guideline.pdf.
16. Bakshi M, Singh S. Development of validated stability-indicating assay methods–critical review. J PharmBiomed Anal [Internet]. 2002. [actualizado 2002; citado 10 Dic 2016]; 28: 1011-1040. Disponible en: http:// www.sciencedirect.com/science/article/pii/S073170850200047X.
17. Norme di Buona Preparazione dei Medicinali in Farmacia. [Internet] Farmacopea Ufficiale. X Edizione; Gennaio 2004. [actualizado Ene 2004; citado 11 Dic 2016] Disponible en: http://www.fog.it/fogliani/giancarlo/normebp.htm.
18. ICH Harmonised Tripartite Guideline. Validation of Analytical Procedures: Text and Methology Q2(R1) [Internet]. 4th Version. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 1994. [actualizado Nov 2005; citado 11 Dic 2016]. Disponible en: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf.
19. Reviewer guidance. Validation of Chromatographic methods. Center for drug evaluation and Research. The Food and Drug administration (FDA).1994.
20. Rekha Madhuri Bai R. On Good Manufacturing Practice in Pharmaceuticals. JOMC. [Internet]. 2016 [citado 11 Dic 2016]; Volume (3); issue 2: 66-71. Disponible en: http://www.rroij.com/open-access/a-review-on-good-manufacturing-practice-in-pharmaceuticals-.pdf.
21. Trissel Lawrence A. Handbook on Injectable Drugs. 16th Edition.Bethesda, Maryland: American Society of Health-System Pharmacists; 2011.
22. Scott E. Walker and Shirley Law. Stability and Compatibility of Granisetron Alone and in Combination with Dexamethasone in 0.9% Sodium Chloride and 5% Dextrose in Water Solutions. Can J Hosp Pharm [Internet]. 2002 [actualizado 2002; citado 11 Dic 2016]; Volume 55:27-38. Disponible en: http://www.cjhp -online.ca/index.php/cjhp/article/download/540/634+&cd=1&hl=es&ct=clnk&gl=es.
23. Moya Gil A, Martínez Gómez MA, Gras Colomer E, Porta Oltra B y Climente Martí M. Stability Studies of Ternary Mixtures Containing Fosaprepitant, Dexamethasone, Ondansetron and Granisetron Used in Clinical Practice. J Anal Bioanal Tech. [Internet]. 2016 [actualizado 2016; citado 11 Dic 2016] Volume 7: 1-7. Disponible en: https://www.omicsonline.org/open-access/ stability-studies-of-ternary-mixtures-containing-fosaprepitantdexamethasone-ondansetron-and-granisetron-used-in-clinical-practice-2155-9872-1000307.pdf.
24. Fundación Hospital Calahorra. Fundación Hospital de Calahorra está implantando desde 2004 el Lean como estrategia para mejora de los procesos. [Internet] 2009. [actualizado 2009; citado 12 Dic 2016] Disponible en: http://www.fhcalahorra.com/noticias/43-fundacion-hospital-calahorra-esta-implantando-desde-2004-el-lean-como-estrategia-para-la-mejora-de-los-procesos.
25. ManMohan S. Sodhi and Navdeep S. Sodhi. Fix the Handful of U.S. Hospitals Responsible for Out-of-Control Costs.[Internet]. Harvar Busines Review. 2013. [actualizado Nov 2013; citado 12 Dic 2016] Disponible en: https://hbr.org/2013/11/fix-the-handful-of-u-s-hospitals-responsible-for-out-of-control-costs/.
26. Font Noguera I. Optimización de procesos en el Hospital La Fe mediante Lean Seis Sigma. [Internet]. Comunicación oral. 59º Congreso Nacional de la Sociedad Española de Farmacia Hospitalaria. Valladolid. 2014. [actualizado 2014; citado 12 Dic 2016] Disponible en: http://www.sefh.es/ sefhpublicaciones/congreso59_ver_ponencias.php?dia=03&horainicio=09&horafin=1010&sala=SS.
27. Real J.M. Detección de oportunidades de mejora mediante la implantación de la metodología Lean. [Internet]. Comunicación oral. 59º Congreso Nacional de la Sociedad Española de Farmacia Hospitalaria. Valladolid. 2014. [actualizado 2014; citado 12 Dic 2016] Disponible en: http://www.sefh.es/sefhpublicaciones/congreso59_ver_ponencias.php?dia=03&horainicio=09&horafin=1010&sala=SS.
28. Real J.M. Detección de oportunidades de mejora mediante la implantación de la metodología Lean. [Internet]. Comunicación oral. 59º Congreso Nacional de la Sociedad Española de Farmacia Hospitalaria. Valladolid. 2014. [actualizado 2014; citado 12 Dic 2016] Disponible en: http://www.sefh.es/sefhpublicaciones/congreso59_ver_ponencias.php?dia=03&horainicio=09&horafin=1010&sala=SS.
____
Descargar artículo en PDF: Estudio de estabilidad de dexametasona como herramienta Lean para la mejora del proceso farmacoterapéutico del paciente onco-hematológico