Rev. OFIL 2017, 27;1:96-98
Fecha de recepción: 22/12/2015 – Fecha de aceptación: 27/10/2016
___
Caro-Teller JM, Alioto D, Serrano-Garrote O, Ferrari-Piquero JM
Hospital Universitario 12 de Octubre. Servicio de Farmacia. Madrid (España)
____
Correspondencia:
José Manuel Caro-Teller
Hospital Universitario 12 de Octubre
(Servicio de Farmacia)
Avenida de Córdoba, s/n
28041Madrid
Correo electrónico: josemanuel.caro@hotmail.com
____
Introducción
La Artritis Idiopática Juvenil Sistémica (AIJS) es una enfermedad inflamatoria reumatológica de origen desconocido que forma parte de un grupo heterogéneo de enfermedades agrupadas bajo el término de Artritis Idiopática Juvenil (AIJ). Supone el 10-20% de los casos de AIJ y es considerada el subtipo de peor pronóstico, ya que dos tercios de la mortalidad de la AIJ se debe a la AIJS1,2.
Los signos y síntomas articulares son las manifestaciones clínicas más típicas. Como manifestaciones extrarticulares se encuentran: exantema evanescente eritematoso, fiebre persistente, linfadenopatía generalizada, hepatoesplenomegalia y anemia. Dichas manifestaciones sistémicas complican la realización de un diagnóstico precoz obligando a realizar al equipo clínico un exhaustivo diagnóstico diferencial3.
El 5-8% de los niños con AIJS desarrollarán Síndrome de Activación Macrofágica (SAM)4. Se trata de una complicación grave con una mortalidad del 8-20%5,6, que debe tratarse urgentemente. Se caracteriza por sangrado espontáneo, fiebre persistente, erupción cutánea, linfadenopatía, hepatoesplenomegalia, ictericia y manifestaciones neurológicas como convulsiones o letargo. En los casos más graves puede instaurarse una coagulopatía intravascular diseminada, fallo multiórganico y coma. Las pruebas de laboratorio revelan pancitopenia, elevación de las enzimas hepáticas, hiperferritinemia, descenso de la velocidad de sedimentación globular en asociación con hipofibrinogenemia por coagulopatía de consumo. La presencia de hemofagocitosis por numerosos macrófagos en el aspirado de médula ósea en ausencia de malignidad es sugerente de SAM6. El tratamiento clásico de SAM secundario a AIJS consiste en bolos de corticoides que pueden asociarse con ciclosporina, mitigando así la tormenta de citocinas que se produce7. En los casos más graves, dada la evidencia disponible, la utilización de un esquema basado en dexametasona, ciclosporina y etóposido durante 8 semanas (protocolo HLH-2004), se convierte en una buena alternativa pese a los riesgos asociados a su uso8.
El tratamiento estándar de la AIJS se basa en combinaciones de AINEs, glucocorticoides y metotrexato. El uso de fármacos biológicos modificadores de la enfermedad estaría relegado a casos refractarios al tratamiento estándar. Tocilizumab (anticuerpo monoclonal anti IL-6) y canakinumab (anticuerpo monoclonal anti IL-1) son los únicos fármacos biológicos indicados para el tratamiento de esta enfermedad9. Canakinumab está indicado para aquellos casos de AIJS mayores de 2 años sin adecuada respuesta al tratamiento previo con AINEs y corticoides sistémicos10, pero debido a su elevado coste y a la escasa experiencia de uso, suele posicionarse como última línea de tratamiento en casos graves y refractarios a tratamientos convencionales previa valoración individualizada.
Descripción del caso
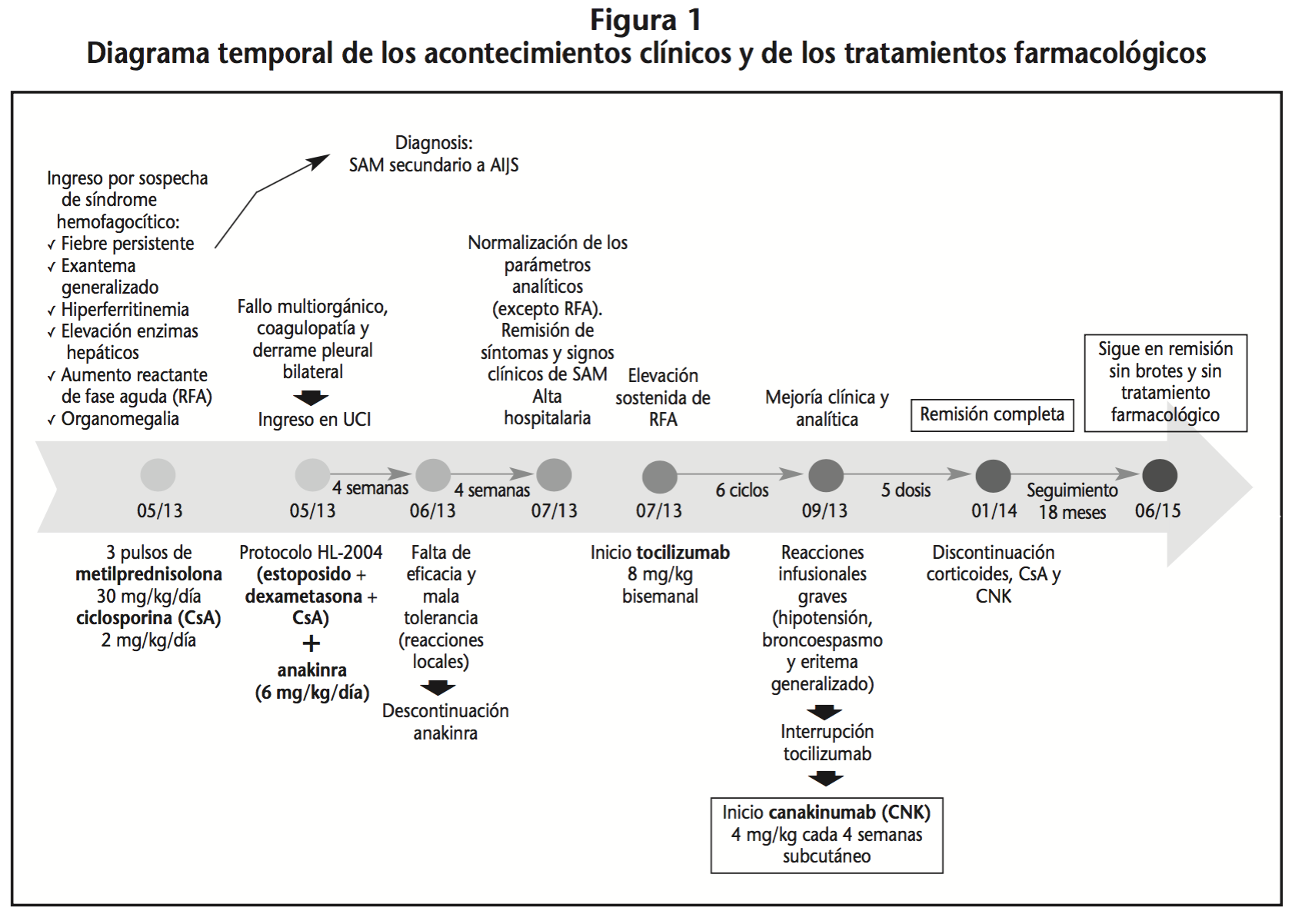
Niña de 8 años que ingresó por sospecha de Síndrome Hemofagocítico. Como principales signos y síntomas presentaba fiebre persistente, exantema generalizado, hiperferritinemia, hipertransaminasemia, reactantes de fase aguda (RFA) disminuidos y organomegalia. Se realizó una biopsia de médula ósea en la que hubo evidencia de hemofagocitosis.
Ante el diagnóstico de SAM inducido por AIJS de simultáneo debut, la paciente fue tratada con tres pulsos de metilprednisolona (30 mg/kg/día) y ciclosporina (2 mg/kg/día). Sus síntomas y signos clínicos empeoraron progresivamente desarrollando fallo multiorgánico, derrame pleural y cuadro hemorrágico por lo que requirió ingreso en la Unidad de Cuidados Intensivos.
Ante la gravedad del caso, se inició el protocolo HLH-2004. Por la refractariedad a corticoides mostrada previamente, se añadió anakinra al tratamiento (6 mg/kg/día), que fue suspendido tras 4 semanas por falta de eficacia (aumento de RFA y persistente colestasis, anemia, neutropenia y trombopenia) y mala tolerabilidad (extremas reacciones infusionales). Al finalizar el protocolo de tratamiento, se logró resolución de la situación con normalización de los parámetros analíticos exceptuando los RFA.
Dada la mejoría clínica la paciente fue dada de alta, pero la elevación sostenida de los RFA llevó a iniciar tratamiento con tocilizumab (8 mg/kg bisemanal) de forma ambulatoria. Con la infusión del quinto ciclo, la paciente desarrolló pápulas no pruriginosas en antebrazos y tronco. Previo a la sexta infusión de tocilizumab, recibió premedicación (dexclorfeniramina y paracetamol) presentando aún así un cuadro de eritema generalizado, broncoespasmo e hipotensión que precisó la administración de adrenalina, esteroides y salbutamol. Visto el beneficio clínico, se planteó realizar un protocolo de desensibilización con tocilizumab pero ante la disponibilidad de canakinumab, el equipo clínico decidió solicitar la autorización de uso como tratamiento alternativo. Pese a no tratarse de un uso compasivo, la Dirección Médica del Hospital solicitó al Centro de Información del Medicamento (Servicio de Farmacia) una valoración del caso. Tras evaluación se decidió realizar un informe favorable calificando como positivo el balance beneficio-riesgo, teniendo en cuenta la ausencia de tratamientos disponibles.
Se inició canakinumab subcutáneo (150 mg cada 28 días) y tras recibir sin incidencias dos ciclos, se objetivó en consulta que la paciente se encontraba afebril, asintomática, sin signos de rash, artralgias ni hepatomegalia. Dada su buena evolución se suspendió el tratamiento con corticoides y ciclosporina. Dos meses después, coincidiendo con la cuarta dosis de canakinumab, se resolvió el cuadro cushingoide y se confirmó la ausencia duradera de síntomas indicativos de enfermedad. La paciente comenzó paulatinamente a realizar sus actividades habituales y volvió al colegio.
El tratamiento con canakinumab se suspendió con su quinta dosis en vista a su buena situación clínica. Tras dieciocho meses de seguimiento, se mantuvo en remisión completa sin brotes y sin precisar tratamiento farmacológico.
Comentario
El SAM es una complicación potencialmente mortal que puede desarrollarse en pacientes con AIJS. En los casos más graves como el presentado, está justificado el tratamiento con ciclosporina y etopósido, aunque la evidencia de uso sea limitada por ser una patología infrecuente8.
Con respecto a la terapia biológica en AIJS, no hay establecido un posicionamiento claro. La utilización de anakinra pese a no tener la indicación aprobada, se basó en presentar un mecanismo de acción muy similar a canakinumab (neutraliza la actividad biológica de la IL-1). Se decidió utilizar tocilizumab previo a canakinumab por ser un fármaco con amplia experiencia de uso.
El posicionamiento favorable del equipo médico y del Servicio de Farmacia a utilizar canakinumab fue decisivo, obteniéndose resultados satisfactorios en cuanto a efectividad y resolviéndose las complicaciones de una enfermedad refractaria a múltiples tratamientos. En cuanto a seguridad, canakinumab fue bien tolerado no apareciendo ninguna de las reacciones adversas descritas en los ensayos clínicos.
Conflicto de intereses: Los autores declaran no tener conflictos de intereses.
Bibliografía
1. Schneider R, Laxer RM. Systemic onset juvenile rheumatoid arthritis. Baillieres Clin Rheumatol. 1998; 12:245-71.
2. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377(9783):2138-49.
3. Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369(9563):767-78.
4. Ravelli A, Magni-Manzoni S, Pistorio A, Besana C, Foti T, Ruperto N, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005; 146(5):598-604.
5. Minoia F, Davì S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66(11):3160-9.
6. Stephan JL, Kone-Paut I, Galambrun C, Mouy R, Bader-Meunier B, Prieur AM. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-92.
7. Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289-98.
8. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-31.
9. NICE evidence summary: new medicine ESNM36. Systemic juvenile idiopathic arthritis:canakinumab. [Acceso Octubre 2016]. Disponible en: https://www. nice.org.uk/advice/esnm36/chapter/key-points-from-the-evidence.
10. Ficha técnica de Ilaris®. [Acceso Octubre 2016]. Disponible en: http://www.ema.europa.eu/docs/es_ES/ document_library/EPAR_-_Product_Information/ human/001109/WC500031680.pdf.
___
Descargar archivo PDF: Experiencia en el uso de canakinumab en el Síndrome de Activación Macrofágica: a propósito de un caso